Epigenetics
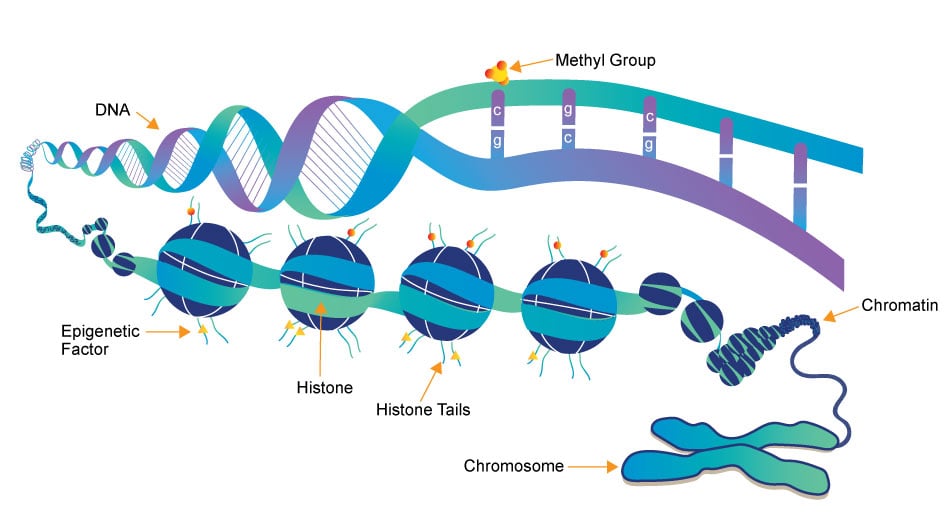
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
PROTAC JAK2 Degrader
SJ1008030 (compound 8) TFA is a selective JAK2-targeting PROTAC that promotes proteasomal degradation of Janus kinase 2 (JAK2). It exhibits potent antiproliferative activity in MHH–CALL-4 leukemia cells, with an IC₅₀ of 5.4 nM. By effectively eliminating JAK2 protein rather than merely inhibiting its activity, SJ1008030 TFA represents a promising therapeutic strategy for JAK2-driven hematologic malignancies, particularly in leukemia research. -
PROTAC BRD4 Degrader
PROTAC BRD4 Degrader-12 (compound 9c) is a bifunctional molecule designed to target and degrade the bromodomain-containing protein BRD4 by recruiting the von Hippel–Lindau (VHL) E3 ubiquitin ligase. It functions as a highly potent degrader, with a DC₅₀ of 0.39 nM and 0.24 nM when conjugated to STEAP1 and CLL1 antibodies, respectively, enabling targeted delivery and degradation of BRD4 in PC3 prostate cancer cells. -
Oligonucleotide Aptamer
AS1411 (AGRO-100) is a G-rich oligonucleotide aptamer that selectively targets nucleolin, a nucleoprotein overexpressed on the surface and within the nucleus of many cancer cells. It inhibits tumor cell proliferation by disrupting nucleolin-containing complexes, thereby affecting key oncogenic processes. AS1411 has been shown to reduce PRMT5 expression and inhibit tumor growth in DU145 prostate cancer cells, and to block the interaction between nucleolin and *bcl-2* mRNA in MCF-7 breast cancer cells, leading to decreased Bcl-2 protein levels and enhanced apoptosis. Beyond its intrinsic anticancer activity, AS1411 serves as a precision-targeting carrier for the delivery of nanoparticles, oligonucleotides, and small molecules to cancer cells. AS1411-conjugated gold nanospheres have demonstrated effective inhibition of breast cancer cell proliferation in vitro and in vivo, with the added advantage of crossing the blood-brain barrier and exhibiting low tissue toxicity, making AS1411 a versatile platform for targeted cancer therapy and drug delivery. -
Aurora B inhibitor
SP-96 is a highly potent, selective, and non-ATP-competitive inhibitor of Aurora B kinase, with an IC₅₀ of 0.316 nM. It exhibits exceptional selectivity, showing over 2000-fold greater specificity for Aurora B compared to off-target kinases such as FLT3 and KIT. In NCI-60 cancer cell line screening, SP-96 demonstrates selective antiproliferative activity, notably against the triple-negative breast cancer (TNBC) cell line MDA-MB-468 (GI₅₀ = 107 nM). SP-96 is a valuable tool for investigating Aurora B–driven oncogenic pathways and holds promise for the development of targeted therapies in TNBC and other malignancies. -
Aurora A inhibitor
CD532 is a potent Aurora A kinase (AURKA) inhibitor with an IC₅₀ of 45 nM. It exerts a dual mechanism of action by both inhibiting AURKA enzymatic activity and promoting the degradation of the MYCN oncoprotein. CD532 directly binds to AURKA and induces a global conformational shift, disrupting its functional interactions. This unique mode of action makes CD532 a valuable tool for cancer research, particularly in MYCN-amplified tumors such as neuroblastoma. -
Multi-target Inhibitor
Chiauranib (CS2164) is an orally active, multi-targeted small molecule inhibitor with potent anticancer activity. It targets key kinases involved in tumor angiogenesis, including VEGFR1, VEGFR2, VEGFR3, PDGFRα, and c-Kit, as well as mitosis-related kinase Aurora B and inflammation-associated kinase CSF-1R. Chiauranib exhibits IC₅₀ values ranging from 1 to 9 nM against these targets. Through simultaneous inhibition of angiogenesis, cell division, and inflammation pathways, Chiauranib exerts strong antitumor effects and is a promising candidate for the treatment of various solid tumors. -
Aurora A inhibitor
JAB-2485 is a highly potent and selective inhibitor of Aurora kinase A (AURKA), with an IC₅₀ of 0.33 nM. It exhibits exceptional selectivity, showing approximately 1700-fold preference for AURKA over Aurora kinase B (AURKB). JAB-2485 induces cell cycle arrest and apoptosis, making it a promising candidate for cancer research, particularly in tumors driven by dysregulated mitotic signaling. -
Aurora A inhibitor
Aurkin A is an allosteric inhibitor that disrupts the interaction between Aurora A kinase (AURKA) and its co-activator TPX2 by selectively targeting the TPX2 binding site on Aurora A. It binds with a dissociation constant (K\_d) of 3.77 μM, thereby interfering with Aurora A activation and its downstream mitotic functions. Aurkin A offers a unique mechanism of action compared to ATP-competitive inhibitors and serves as a valuable tool for studying Aurora A–TPX2–mediated signaling in cell division and cancer progression. -
Aurora A-TPX2 interaction inhibitor
CAM2602 is a selective inhibitor targeting the interaction between Aurora A kinase and its co-activator TPX2, with a high binding affinity of 19 nM for Aurora A. It effectively inhibits the growth of pancreatic cancer cells and demonstrates antitumor activity in solid tumor transplant models. Mechanistically, CAM2602 increases the proportion of phospho-histone H3 (PH3) positive cells—indicative of mitotic arrest—while decreasing the levels of Aurora A phosphorylated at threonine 288 (P-Thr288), a marker of Aurora A activation. These effects collectively contribute to its ability to disrupt mitosis and suppress tumor progression. -
Aurora kinase inhibitor
DBPR728 is an acyl prodrug of 6K465, engineered to improve pharmacokinetic properties by reducing the number of hydrogen bond donors, thereby enhancing membrane permeability and stability. As a prodrug of 6K465, DBPR728 functions as an Aurora kinase inhibitor that destabilizes MYC family oncoproteins, including c-MYC and N-MYC. It exhibits potent antitumor activity, particularly in cancers characterized by MYC overexpression. Notably, DBPR728 offers a 10-fold increase in oral bioavailability compared to 6K465, making it a promising candidate for the development of orally administered therapies targeting MYC-driven malignancies. - Phthalazinone pyrazole is a potent, selective, and orally bioavailable inhibitor of Aurora-A kinase, with an IC₅₀ of 0.031 μM. It exerts antitumor effects by arresting mitosis and inducing apoptosis in proliferating cells, leading to inhibition of tumor growth. Additionally, phthalazinone pyrazole suppresses epithelial-mesenchymal transition (EMT) during the differentiation of hepatocyte-like cells (HLCs) derived from human embryonic stem cells, indicating potential applications in both cancer therapy and stem cell biology.
-
Aurora Kinase A/JAK2 Inhibitor
AJI-214 is a dual-target inhibitor that simultaneously inhibits Aurora kinase A and Janus kinase 2 (JAK2). It directly blocks Aurora A activity, disrupting T cell mitotic progression and polarity, while also inhibiting JAK2-mediated STAT3 phosphorylation, thereby suppressing the differentiation of pro-inflammatory TH1 and TH17 cells. AJI-214 holds therapeutic potential for modulating immune responses and is being investigated for the prevention and treatment of graft-versus-host disease (GVHD). -
Aurora A inhibitor.
6K465 is a pyrimidine-based small molecule inhibitor selectively targeting Aurora A kinase (AURKA). By inhibiting AURKA activity, 6K465 effectively reduces the expression levels of the oncogenic transcription factors c-MYC and N-MYC, contributing to its anticancer effects. This compound shows promise as a therapeutic agent for MYC-driven cancers by disrupting mitotic regulation and oncogene stabilization. -
Aurora kinase inhibitor
Derrone is a prenylated isoflavone that functions as an Aurora kinase inhibitor, exhibiting IC₅₀ values of 6 μM for Aurora B and 22.3 μM for Aurora A. By targeting these key mitotic kinases, Derrone disrupts cell cycle progression and displays notable antitumor activity, making it a promising compound for cancer research focused on mitotic regulation. -
HDAC11 inhibitor
Elevenostat (JB3-22) is a selective histone deacetylase 11 (HDAC11) inhibitor with an IC₅₀ of 0.235 µM. It exhibits antitumor activity by inducing apoptosis in multiple myeloma cells and shows potential as a therapeutic agent in hematologic malignancies. Additionally, Elevenostat has been shown to inhibit the maturation of mouse oocytes, suggesting a role for HDAC11 in reproductive biology and offering a tool for studying epigenetic regulation in oocyte development. -
HDAC6/HDAC10/LTA4H Inhibitor
Bufexamac is a nonsteroidal anti-inflammatory drug (NSAID) that functions as a dual inhibitor of class IIb histone deacetylases—HDAC6 and HDAC10—as well as leukotriene A4 hydrolase (LTA4H). It exhibits binding affinities (K\_d) of 0.53 µM for HDAC6 and 0.22 µM for HDAC10. Through its dual inhibitory activity, Bufexamac combines epigenetic modulation with anti-inflammatory effects, making it a valuable compound for research in inflammation, immune regulation, and HDAC-related pathologies. -
HDAC6 inhibitor
TYA-018 is an orally active, potent, and highly selective inhibitor of histone deacetylase 6 (HDAC6). In preclinical studies, TYA-018 demonstrates cardioprotective effects by preserving heart function in mice. Additionally, it enhances systemic energetics by upregulating the expression of genes involved in fatty acid metabolism, protein turnover, and oxidative phosphorylation, highlighting its potential in both cardiovascular and metabolic disease research. -
Snail/HDAC inhibitor
CYD19 is a potent dual-target inhibitor that simultaneously disrupts Snail and HDAC1 activity. It exhibits an IC₅₀ of 0.405 μM against HDAC1 and binds Snail with a K\_d of 0.18 μM. In HCT-116 colorectal cancer cells, CYD19 increases histone H4 acetylation and downregulates Snail protein expression, leading to the induction of apoptosis. This dual mechanism positions CYD19 as a promising therapeutic candidate for targeting epithelial–mesenchymal transition (EMT) and epigenetic dysregulation in cancer. -
CoreDAC Inhibitor
TNG260 is a selective and orally bioavailable inhibitor of histone deacetylase 1 (HDAC1) and the CoREST transcriptional corepressor complex. It exhibits approximately 10-fold selectivity for HDAC1 over HDAC3 and 500-fold selectivity for the CoREST complex compared to other HDAC-containing complexes such as NuRD and Sin3. TNG260 modulates the tumor immune microenvironment by reducing immunosuppressive neutrophil infiltration, enhancing effector T cell recruitment, and reversing anti-PD-1 resistance associated with STK11 mutations through inhibition of the CoREST–HDAC1 axis. In preclinical models, TNG260 induces durable tumor regression when combined with α-PD-1 therapy in MC38 tumor-bearing mice harboring STK11 mutations, while demonstrating reduced hematologic toxicity compared to non-selective HDAC inhibitors. -
phospholipase A2/HDAC2 inhibitor
Rhamnetin is a naturally occurring flavonoid and quercetin derivative found in *Coriandrum sativum*. It functions as an inhibitor of secretory phospholipase A₂ and histone deacetylase 2 (HDAC2), contributing to its broad pharmacological profile. Rhamnetin exhibits notable antitumor, antioxidant, and anti-inflammatory activities, making it a promising compound for research in cancer, oxidative stress-related conditions, and inflammatory diseases. -
HDAC10 inhibitor
DKFZ-748 is a selective histone deacetylase 10 (HDAC10) inhibitor with a pIC₅₀ of 7.66, corresponding to an IC₅₀ of approximately 22 nM. It exhibits antitumor activity, making it a valuable tool for studying HDAC10-mediated biological processes and a promising candidate for the development of targeted cancer therapies. -
HDAC6 inhibitor
KA2507 is a potent, orally active, and highly selective histone deacetylase 6 (HDAC6) inhibitor with an IC₅₀ of 2.5 nM. It exhibits strong antitumor activity and immunomodulatory effects in preclinical models, making it a promising candidate for cancer therapy, particularly in tumors where HDAC6 plays a key role in tumor progression and immune evasion. -
GRK5 inhibitor
KR-39038 is a potent and orally bioavailable inhibitor of G protein-coupled receptor kinase 5 (GRK5), with an IC₅₀ of 0.02 μM. It effectively suppresses angiotensin II–induced cellular hypertrophy by inhibiting the HDAC5 signaling pathway in neonatal cardiomyocytes. KR-39038 exhibits strong anti-hypertrophic activity and improves cardiac function in preclinical models, making it a promising candidate for research in heart failure and related cardiovascular diseases. -
HDAC6 inhibitor
T-518 is an orally active, selective histone deacetylase 6 (HDAC6) inhibitor with an IC₅₀ of 36 nM for human HDAC6. It is capable of crossing the blood–brain barrier, making it suitable for central nervous system applications. T-518 is particularly useful in tauopathy research, where HDAC6 inhibition may modulate tau pathology and neurodegenerative processes. -
HDAC1 inhibitor
9-Hydroxyoctadecanoic acid (9-HSA) is a naturally derived inhibitor of histone deacetylase 1 (HDAC1), inhibiting approximately 66.4% of HDAC1 enzymatic activity at a concentration of 5 μM. It exhibits notable anticancer activity, likely through epigenetic modulation of gene expression, making it a promising compound for cancer research and therapeutic development targeting HDAC1-regulated pathways. -
HDAC6 inhibitor
SE-7552 is an orally active, highly selective histone deacetylase 6 (HDAC6) inhibitor, based on a 2-(difluoromethyl)-1,3,4-oxadiazole (DFMO) scaffold, with an IC₅₀ of 33 nM. As a non-hydroxamate HDAC6 inhibitor, SE-7552 demonstrates exceptional isoform selectivity, showing over 850-fold preference for HDAC6 compared to other HDAC isozymes. In preclinical studies, SE-7552 effectively inhibits multiple myeloma tumor growth in vivo and also exhibits anti-obesity effects in diet-induced obese mouse models, highlighting its therapeutic potential in both oncology and metabolic disease research. -
COX-1/HDAC/Tyrosinase Inhibitor
Gnetol is a bioactive phenolic compound isolated from the root of *Gnetum montanum* with diverse pharmacological properties. It potently inhibits cyclooxygenase-1 (COX-1) with an IC₅₀ of 0.78 μM and exhibits histone deacetylase (HDAC) inhibitory activity. Gnetol is also a strong tyrosinase inhibitor, with an IC₅₀ of 4.5 μM against murine tyrosinase, leading to suppression of melanin biosynthesis. In addition to its antioxidant, antiproliferative, anticancer, and hepatoprotective effects, Gnetol modulates metabolic enzymes in a concentration-dependent manner, including α-amylase, α-glucosidase, and adipogenesis pathways, making it a promising candidate for research in oncology, dermatology, and metabolic disorders. -
HDAC inhibitor
Marein is a natural compound with multifaceted pharmacological properties, including HDAC inhibition with an IC₅₀ of 100 μM. It exerts neuroprotective effects by preserving mitochondrial function and activating the AMPK signaling pathway. In HepG2 cells, Marein improves high glucose–induced insulin resistance by enhancing glucose uptake via the CaMKK/AMPK/GLUT1 pathway, promoting glycogen synthesis through the IRS/Akt/GSK-3β pathway, and suppressing gluconeogenesis via the Akt/FoxO1 axis. Additionally, Marein possesses antioxidative, antihypertensive, antihyperlipidemic, and antidiabetic properties, making it a promising candidate for metabolic and neurodegenerative disease research. -
HDAC inhibitor
Crebinostat is a potent, broad-spectrum histone deacetylase (HDAC) inhibitor with IC₅₀ values of 0.7 nM (HDAC1), 1.0 nM (HDAC2), 2.0 nM (HDAC3), and 9.3 nM (HDAC6). It effectively induces acetylation of histone H3 and H4, and enhances the expression of Egr1, a cAMP response element-binding protein (CREB) target gene. In cultured neurons, Crebinostat increases the density of synapsin-1 puncta along dendrites, indicating enhanced synaptic connectivity. By modulating chromatin-mediated neuroplasticity, Crebinostat has been shown to improve memory performance in mice, supporting its potential for neuropsychiatric and cognitive disorder research. -
HDAC inhibitor
Bakkenolide A is a natural sesquiterpene lactone isolated from *Petasites tricholobus*. It exhibits anti-leukemic activity by modulating epigenetic and signaling pathways, specifically through inhibition of histone deacetylase 3 (HDAC3) and regulation of the PI3K/Akt signaling cascade. These combined effects contribute to its potential as a therapeutic agent in leukemia research. -
HDAC8 inhibitor
NCC-149 is a selective inhibitor of histone deacetylase 8 (HDAC8), making it a valuable tool for studying HDAC8-specific functions. It has shown utility in promoting neural differentiation, and is particularly useful in research focused on neurodevelopmental processes and potential therapeutic strategies for neurological disorders involving epigenetic dysregulation. -
HDAC inhibitor
CM-1758 is a histone deacetylase (HDAC) inhibitor with demonstrated antitumor activity in vivo. In acute myeloid leukemia (AML) cells, CM-1758 induces the acetylation of non-histone proteins, suggesting a broader epigenetic and post-translational regulatory effect beyond chromatin remodeling. Its dual impact on tumor growth and protein acetylation supports its potential as a therapeutic agent in hematologic malignancies. -
HDAC6 inhibitor
MPT0G211 is a highly potent, orally bioavailable, and selective histone deacetylase 6 (HDAC6) inhibitor, with an IC₅₀ of 0.291 nM and over 1000-fold selectivity against other HDAC isoforms. It efficiently crosses the blood–brain barrier and demonstrates neuroprotective effects by reducing tau phosphorylation and improving cognitive function in Alzheimer's disease models. Additionally, MPT0G211 exhibits anti-metastatic and anticancer activities, making it a promising therapeutic candidate for both neurodegenerative disorders and oncology research. -
HDAC inhibitor
MPT0B390 is a potent arylsulfonamide-based histone deacetylase (HDAC) inhibitor that also functions as an inducer of tissue inhibitor of metalloproteinases 3 (TIMP3). It exhibits strong antitumor properties, including inhibition of tumor growth, metastasis, and angiogenesis. MPT0B390 shows significant antiproliferative activity against the human colon cancer cell line HCT116, with a GI₅₀ of 0.03 μM, highlighting its potential as a promising candidate for cancer therapy. -
Endogenous Metabolite
Triacetin (Glyceryl triacetate) is an orally active synthetic triester of glycerol and acetic acid that serves as a bioavailable source of acetate. It freely crosses the blood–brain barrier and cellular membranes, making it particularly effective in targeting central nervous system malignancies. In glioma cells, Triacetin increases intracellular acetate levels, promotes histone acetylation, and induces cell cycle arrest and apoptosis. Additionally, Triacetin enhances the chemotherapeutic efficacy of Temozolomide (TMZ), supporting its potential as an adjuvant in glioma treatment strategies. -
class-IIa HDAC inhibitor
NT160 is a highly potent inhibitor of class IIa histone deacetylases (HDACs), with an IC₅₀ value of 0.046 μM. Due to its strong inhibitory activity and potential central nervous system (CNS) penetration, NT160 is a valuable compound for investigating the role of class IIa HDACs in CNS-related disorders, including neurodegenerative and neuropsychiatric diseases. -
DNA Methyltransferase Inhibitor
RSC133 is a dual epigenetic modulator that inhibits both histone deacetylases (HDACs) and DNA methyltransferases (DNMTs). It effectively enhances the reprogramming of human somatic cells into induced pluripotent stem cells (iPSCs) and supports the maintenance of pluripotency by promoting an undifferentiated state. RSC133 is a valuable tool for stem cell research and epigenetic reprogramming studies. -
HDAC inhibitor
Alteminostat (CKD-581) is a potent, broad-spectrum histone deacetylase (HDAC) inhibitor that targets both class I and class II HDAC isoforms. It promotes histone H3 and α-tubulin acetylation, indicating effective inhibition of nuclear and cytoplasmic HDAC activity. Alteminostat is being investigated for its therapeutic potential in hematologic malignancies, including lymphoma and multiple myeloma, and serves as a valuable tool in epigenetic cancer research. -
HDAC6 inhibitor
CG347B is a selective inhibitor of histone deacetylase 6 (HDAC6) and also serves as a structural scaffold in the synthesis of other metalloenzyme inhibitors. Due to its HDAC6 selectivity, CG347B is a valuable tool for research in oncology, immunology, and neurology, where HDAC6 plays key roles in regulating protein homeostasis, immune response, and neurodegenerative processes. -
HDAC inhibitor
MC1742 is a potent pan-HDAC inhibitor with broad activity across multiple HDAC isoforms, exhibiting IC₅₀ values of 0.1 μM (HDAC1), 0.11 μM (HDAC2), 0.02 μM (HDAC3), 0.007 μM (HDAC6), 0.61 μM (HDAC8), 0.04 μM (HDAC10), and 0.1 μM (HDAC11). It effectively increases acetylation of histone H3 and α-tubulin, markers of HDAC inhibition. MC1742 inhibits the growth of cancer stem cells (CSCs), and induces growth arrest, apoptosis, and differentiation, particularly in sarcoma CSC models, making it a promising candidate for targeting therapy-resistant cancer cell populations. -
HDAC1 inhibitor
SB-429201 is a potent and selective inhibitor of histone deacetylase 1 (HDAC1), with an IC₅₀ of approximately 1.5 μM. It exhibits at least a 20-fold selectivity for HDAC1 over other class I isoforms, including HDAC3 and HDAC8. SB-429201 serves as a valuable tool for studying HDAC1-specific functions and may have potential applications in epigenetic and cancer research. - Oleuropein Aglycone (3,4-DHPEA-EA) is a bioactive polyphenol and the aglycone form of oleuropein, generated through enzymatic, acidic, or acetylated hydrolysis. It exhibits a broad range of pharmacological effects. In a TgCRND8 transgenic mouse model of Alzheimer’s disease, dietary supplementation (50 mg/kg) increases neuronal autophagic vesicles, reverses cognitive deficits, and reduces histone deacetylase 2 (HDAC2) levels in the cortex and hippocampus. In a high-fat diet-induced obesity rat model, Oleuropein Aglycone elevates urinary norepinephrine, interscapular brown adipose tissue epinephrine, and UCP1 protein levels, while reducing plasma leptin levels and total abdominal fat mass. Additionally, in a carrageenan-induced pleurisy mouse model, it mitigates lung neutrophil infiltration, lipid peroxidation, and IL-1β production. These findings highlight its potential in neurodegenerative, metabolic, and inflammatory disease research.
-
HDAC1/HDAC2 inhibitor
BRD2492 (compound 6d) is a potent and selective inhibitor of histone deacetylases HDAC1 and HDAC2, with IC₅₀ values of 13.2 nM and 77.2 nM, respectively. It exhibits over 100-fold selectivity for HDAC1/2 compared to HDAC3 and HDAC6. BRD2492 effectively inhibits the proliferation of breast cancer cell lines, with IC₅₀ values of 1.01 μM for T-47D cells and 11.13 μM for MCF-7 cells, highlighting its potential as a targeted epigenetic therapeutic in breast cancer research. -
HDAC8 inhibitor
1-Naphthohydroxamic acid (Compound 2) is a potent and selective inhibitor of histone deacetylase 8 (HDAC8), with an IC₅₀ of 14 μM. It demonstrates high selectivity for HDAC8 over other HDAC isoforms, showing minimal activity against class I HDAC1 and class II HDAC6 (IC₅₀ > 100 μM). Unlike broad-spectrum HDAC inhibitors, 1-Naphthohydroxamic acid does not increase global histone H4 acetylation or reduce total intracellular HDAC activity, but it effectively induces tubulin acetylation. This selective profile makes it a valuable tool for studying HDAC8-specific functions. -
HDAC inhibitor
YSR734 (Compound 21) is a covalent histone deacetylase (HDAC) inhibitor with IC₅₀ values of 110 nM, 154 nM, and 143 nM for HDAC1, HDAC2, and HDAC3, respectively. It induces apoptosis in leukemia cells and promotes myoblast differentiation, making it a valuable compound for both cancer research and studies related to muscle regeneration. YSR734 is particularly relevant in the investigation of therapeutic strategies for Duchenne muscular dystrophy. -
HDAC6 inhibitor
PB131 is a highly selective and brain-permeable histone deacetylase 6 (HDAC6) inhibitor, exhibiting strong binding affinity with an IC₅₀ of 1.8 nM. It possesses potent anti-inflammatory activity and is particularly suited for research in inflammation-related disorders, including neuroinflammation, due to its effective central nervous system penetration and HDAC6 specificity. -
HDAC2/HDAC3 inhibitor
MI-192 is a selective inhibitor of histone deacetylases HDAC2 and HDAC3, with IC₅₀ values of 30 nM and 16 nM, respectively. It demonstrates high selectivity for HDAC2/3 over other HDAC isoforms. MI-192 induces apoptosis in myeloid leukemia cells and exhibits both anticancer and neuroprotective activities, making it a valuable compound for research in oncology and neurodegenerative diseases. -
HDAC inhibitor
Pivanex (AN-9) is an orally active histone deacetylase (HDAC) inhibitor derived from butyric acid. It downregulates BCR-ABL protein expression and promotes apoptosis, contributing to its antitumor effects. In addition, Pivanex exhibits antimetastatic and antiangiogenic properties, making it a promising candidate for research in hematologic malignancies and solid tumors. -
HDAC inhibitor
BRD4884 is a potent histone deacetylase (HDAC) inhibitor that selectively targets class I HDACs. It exhibits IC₅₀ values of 29 nM for HDAC1, 62 nM for HDAC2, and 1.09 µM for HDAC3. Its differential potency across HDAC isoforms makes BRD4884 a valuable tool for investigating HDAC1/2-mediated epigenetic regulation and their roles in disease pathogenesis. -
HDAC inhibitor
FNDR-20123 is a first-in-class, orally active histone deacetylase (HDAC) inhibitor developed for antimalarial therapy. It demonstrates potent inhibitory activity against both Plasmodium and human HDACs, with IC₅₀ values of 31 nM and 3 nM, respectively. FNDR-20123 effectively targets multiple stages of *Plasmodium falciparum*, with IC₅₀ values of 41 nM for the asexual blood stage and 190 nM for male gametocytes. It inhibits HDAC1, HDAC2, HDAC3, HDAC6, and HDAC8 with IC₅₀ values of 25, 29, 2, 11, and 282 nM, respectively, and also exhibits nanomolar-level inhibition of Class III HDAC isoforms. FNDR-20123 shows a favorable safety profile, supporting its potential as a novel antimalarial agent targeting epigenetic regulation.
