Epigenetics
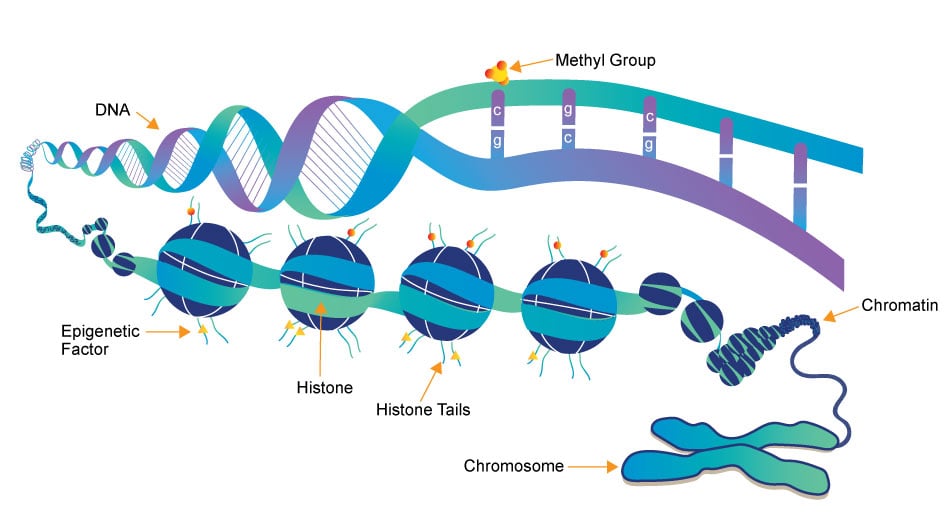
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
Aurora A Inhibitor
CD532 hydrochloride is a selective inhibitor of Aurora A kinase, exhibiting an IC50 value of 45 nM. This compound not only inhibits Aurora A activity but also promotes the degradation of MYCN. Additionally, CD532 hydrochloride directly interacts with AURKA, leading to a significant conformational change. This reagent is valuable for cancer research, particularly in studies focusing on cell proliferation and tumor progression. -
Aurora Kinase Inhibitor
Aurora kinase inhibitor-2 is a selective, ATP-competitive inhibitor targeting Aurora kinases A and B, exhibiting IC50 values of 310 nM and 240 nM, respectively. This compound is valuable for studying cell cycle regulation and mitosis, making it suitable for research applications related to cancer biology and therapeutic development. Its precision in inhibiting aurora kinase activity allows for further exploration of signaling pathways associated with tumor growth and progression. -
Aurora-A/Aurora-B PROTAC Degrader
dAurAB2 is a dual-targeting PROTAC designed to degrade Aurora-A and Aurora-B, demonstrating potent efficacy with DC50 values of 59 nM and 39 nM, respectively. This compound effectively reduces N-Myc protein levels in MYCN-amplified IMR32 neuroblastoma cells, making it a valuable tool for neuroblastoma research. The unique design incorporates a specific Aurora ligand and an E3 ligase ligand connected by a tailored linker, facilitating targeted degradation and advancing studies in cancer biology. -
Aurora-A Inhibitor
Aurora-A ligand 1 is a specific inhibitor of Aurora-A, exhibiting a high-affinity binding with a dissociation constant (Kd) of 0.85 nM. It serves as a crucial ligand for the development of PROTAC-based Aurora-A degraders, contributing to anti-tumor activity. Additionally, Aurora-A ligand 1 can be utilized in the synthesis of HLB-0532259, which has demonstrated potent anti-tumor effects against neuroblastoma, highlighting its potential in cancer research. -
MASTL/Aurora A Kinase Dual Inhibitor
MASTL/Aurora A-IN-1 is a dual inhibitor of MASTL and Aurora A kinases, exhibiting IC50 values of 0.56 μM and 0.16 μM, respectively. This compound demonstrates broad-spectrum anticancer activity, showing potent effects against various cell lines, including SR, K-562, MDA-MB-435, MOLT-4, and SK-MEL-2, with GI50 values ranging from 0.023 to 0.051 μM. By inhibiting these kinases, MASTL/Aurora A-IN-1 induces G2/M cell cycle arrest, thereby effectively inhibiting cancer cell proliferation. Its application is particularly valuable in cancer research, especially for studying tumors with dysregulated mitosis. -
Aurora Kinase Inhibitor
Aurora Kinase Inhibitor-8 selectively targets Aurora kinases, which play critical roles in mitotic regulation and are frequently implicated in tumorigenesis. This compound exhibits potent inhibitory activity, making it a valuable tool for studying the cell cycle and cancer biology. Research applications include elucidating the mechanisms of cell proliferation and exploring therapeutic strategies for cancer treatment. -
Aurora A kinase Inhibitor
Aurora kinase inhibitor-13 is a selective inhibitor of Aurora A kinase, exhibiting an IC50 value of 2.3 μM. This compound effectively disrupts the function of Aurora A, a key regulator of cell cycle progression. Its ability to modulate Aurora kinase activity makes it valuable for research focused on cancer biology and the development of targeted therapeutics. -
Aurora Kinase A Inhibitor
Aurora kinase-IN-4 is a covalent and ATP-competitive inhibitor of Aurora Kinase A, exhibiting an IC50 of 1.7 nM. This compound demonstrates significant activity in inhibiting cell proliferation across various cancer cell lines, including SJSA-1, MDA-MB-231, A54, and HeLa, with IC50 values of 4.27, 1.54, 3.08, and 6.99 μM, respectively. Aurora kinase-IN-4 is particularly relevant for research into triple-negative breast cancer (TNBC), making it a valuable tool for studies in oncology. -
Aurora A PROTAC Degrader
AurAP14 is a PROTAC degrader specifically targeting Aurora A, with a DC50 of 120 nM. This compound exhibits potent inhibitory effects on various tumor cell lines, showing IC50 values of 0.294 μM in A549 cells and 0.534 μM in MCF-7 cells. AurAP14 induces apoptosis while effectively arresting A549 cells in the S and G2/M phases of the cell cycle. Additionally, AurAP14 demonstrates significant anti-tumor efficacy in nude mouse xenograft models of A549 and A549/PTR, making it a valuable tool for research focused on treating Aurora A-overexpressing non-small cell lung cancer (NSCLC). -
CDK4/6/9-AURKA/B Inhibitor
LCI133 is a selective multikinase inhibitor targeting CDK4, CDK6, CDK9, and AURKA/B, exhibiting nanomolar potency (IC50 values of 4.7 nM, 10.2 nM, 4.1 nM, 2.8 nM, and 10.6 nM, respectively). It effectively induces S/G2 cell-cycle arrest and promotes significant apoptosis in MYCN-amplified neuroblastoma BE(2)-C cells. Additionally, LCI133 demonstrates notable antitumor efficacy in preclinical models, particularly in BE(2)-C neuroblastoma xenograft studies, making it a valuable tool for cancer research and therapeutic development. -
Aurora kinase A/B Inhibitor
IBPR002 is a potent inhibitor of Aurora kinase A and B, exhibiting IC50 values of 41 nM and 17 nM, respectively. This compound disrupts the nucleation and bundling of kinetochore microtubules, impairs the bipolarity of mitotic spindles, and enhances the binding of non-phosphorylated hepatoma up-regulated protein (HURP) to mother centrosome-derived microtubules. IBPR002 demonstrates significant anti-tumor activity in a colorectal cancer xenograft model, making it valuable for research focused on colorectal cancer mechanisms and therapeutics. -
Aurora Kinase Inhibitor
AKI-001 is a potent inhibitor of Aurora kinases, specifically targeting Aurora A and Aurora B with an IC50 of less than 100 nM. This pentacyclic compound demonstrates significant cellular efficacy, making it a valuable tool for investigating cell cycle regulation and mitotic progression. Its selective inhibitory action positions AKI-001 as an essential reagent for research in cancer biology and therapeutic development. -
Aurora A/PKC Inhibitor
(Rac)-Aurora A/PKC-IN-1 is a potent inhibitor of Aurora A and protein kinase C (PKC) isoforms α, β1, β2, and θ. This compound demonstrates significant antiproliferative effects in breast cancer cell lines in vitro and exhibits antimetastatic properties in vivo. It serves as a valuable tool for researchers investigating the role of these kinases in cancer biology and therapeutic strategies. -
BET/Aurora kinase Inhibitor
BET/Aurora kinase-IN-1 is a dual inhibitor targeting both BET and Aurora kinases. This compound demonstrates significant antiproliferative activity across various cancer cell lines and exhibits notable antitumor efficacy in xenograft models of renal cell cancer and colon cancer, achieving tumor growth inhibition rates of 45.99% and 53.06%, respectively. BET/Aurora kinase-IN-1 is a valuable tool for researchers investigating cancer biology and therapeutic strategies targeting these kinases. -
Aurora Kinase Inhibitor
SNS-314 is a potent and selective inhibitor of aurora kinases, demonstrating IC50 values of 9 nM for Aurora A, 31 nM for Aurora B, and 6 nM for Aurora C. This compound effectively disrupts mitotic processes and is valuable in cancer research for studying cell cycle regulation and tumor growth inhibition. SNS-314 is particularly useful for investigations into therapies targeting aurora kinases in various malignancies. -
Aurora A Inhibitor
MLN8054 sodium is a selective inhibitor of Aurora A kinase, which plays a critical role in cell cycle regulation. This compound enhances radiosensitivity and can activate DNA double-strand break responses in prostate cancer cells during in vitro assays. Its mechanism induces accumulation of cells in the G2/M phase and promotes polyploidy. In vivo studies demonstrate that MLN8054 sodium significantly delays tumor growth and enhances apoptosis in cancer cells when administered alongside radiotherapy, making it a valuable tool for cancer research and treatment strategies. -
Aurora B Inhibitor
Aurora kinase inhibitor-10 is a potent inhibitor of Aurora B with an IC50 of 8 nM. This small molecule demonstrates significant antitumor activity, making it a valuable tool for cancer research. Its ability to selectively target Aurora B kinase supports investigations into mitotic regulation and offers potential therapeutic insights for tumor treatments. -
Aurora-A Ligand
Aurora-A ligand 2 is a selective ligand for the Aurora-A kinase, functioning as a crucial component in PROTAC technology. It plays a significant role in the targeted degradation of Aurora-A, facilitating the investigation of its biological implications in cancer research. This compound is valuable for studying Aurora-A kinases and exploring therapeutic strategies in oncology. -
Aurora Kinase Inhibitor
Aurora kinase inhibitor-11 is a potent inhibitor of Aurora Kinase, exhibiting an IC50 of 0.14 μM. This compound demonstrates significant anticancer activity, making it a valuable tool for research applications focused on cancer biology and therapeutic strategies targeting mitotic processes. Its efficacy in modulating kinase activity positions it as a relevant candidate for studies aimed at understanding tumorigenesis and developing novel cancer treatments. -
Aurora Kinase Inhibitor
Tripolin A is a selective non-ATP competitive inhibitor of Aurora A kinase, exhibiting IC50 values of 1.5 μM for Aurora A and 7 μM for Aurora B. This compound plays a crucial role in modulating cell cycle progression by targeting Aurora kinases, making it valuable in cancer research. Tripolin A is used to investigate the mechanisms of mitotic regulation and potential therapeutic strategies in tumor cells. -
Aurora Kinase Inhibitor
XMD-12 is a selective Aurora kinase inhibitor that demonstrates significant anti-tumor activity. It effectively enhances paclitaxel-induced cell death and exhibits high potency against Aurora A, B, and C kinases, with IC50 values of 5.6, 18.4, and 24.6 nM, respectively. This compound is valuable for research applications in cancer biology and therapy development. -
Aurora Kinases Inhibitor
Aurora kinase-IN-2 is a potent inhibitor of Aurora kinases, demonstrating IC50 values of 90 nM for Aurora A and 152 nM for Aurora B. This compound effectively induces cell cycle arrest at the G2/M phase by modulating cyclin B1 and cdc2. It is primarily utilized in cancer research to explore the role of Aurora kinases in tumorigenesis and therapeutic response. -
Aurora A Inhibitor
Aurora A Inhibitor 1 is a potent and selective inhibitor of the Aurora A kinase, which plays a crucial role in regulating cell division and has been implicated in various cancers. Overexpression of Aurora A is associated with oncogenic properties, making it an important target for cancer research. This compound is suitable for studies focusing on the therapeutic modulation of Aurora A in diverse cancer types. -
Aurora Kinase Inhibitor
Aurora kinase inhibitor-9 is a potent dual inhibitor of Aurora A and Aurora B kinases, exhibiting IC50 values of 0.093 µM and 0.09 µM, respectively. This compound demonstrates significant anti-proliferative activity across various cancer cell lines, making it a valuable tool in cancer research. Its ability to target key regulators of cell division positions it as a candidate for studies investigating mitotic disruption and the development of novel cancer therapies. -
Aurora Inhibitor
TAK-901 hydrochloride is a potent inhibitor of aurora kinases A and B, exhibiting IC50 values of 21 nM and 15 nM, respectively. This compound disrupts cell cycle progression, making it a valuable tool in cancer research and therapeutic development. Its ability to inhibit aurora kinases positions TAK-901 hydrochloride as an important reagent for studying mitotic regulation and exploring targeted cancer therapies. -
Aurora A PROTAC Degrader
PROTAC Aurora A Degrader-1 is a selective degrader that targets Aurora A, effectively forming a ternary complex with AURKA and CRBN. This compound demonstrates potent biological activity, inducing degradation of AURKA, lowering MYCN levels, and promoting DNA damage and apoptosis in cancer cells. With DC50 values of 1 nM and 2 nM in LAN5 and SMS-SAN cells, respectively, it exhibits significant antiproliferative effects and is valuable for research on neuroblastoma and small cell lung cancer. -
Aurora Kinase Inhibitor
Tripolin B is an ATP-competitive inhibitor targeting Aurora kinases, exhibiting IC50 values of 2.5 µM and 6 µM for Aurora A and Aurora B kinases, respectively. This compound has demonstrated selectivity in its inhibition profile and is primarily utilized in cellular studies to explore the roles of Aurora kinases in cell cycle regulation and cancer progression. Tripolin B can be a valuable tool in research focused on cell division and oncogenic signaling pathways. -
Aurora Kinase Inhibitor
BI 831266 is a potent and selective inhibitor of Aurora kinase B, a critical regulator of mitosis. This compound exhibits significant antitumor activity, making it valuable for cancer research. Its inhibition of Aurora B can lead to disruptions in cell division, providing insights into potential therapeutic applications for various malignancies. -
Aurora A/B Kinases Inhibitor
TY-011 is a selective inhibitor of Aurora A and B kinases, disrupting normal microtubule-kinetochore attachment. This interference results in DNA damage and apoptosis, effectively inhibiting the proliferation of human gastric cancer cells, with observed IC50 values between 0.11 and 4.49 μM across various gastric cancer cell lines. TY-011 serves as a valuable tool in the study of gastric cancer and the mechanisms underlying mitotic regulation. -
Aurora Kinase Inhibitor
OM137 is a potent Aurora Kinase inhibitor, demonstrating IC50 values of 21.7 μM for Aurora A kinase and 2.4 μM for Aurora B kinase. Additionally, OM137 affects cell cycle regulation by inhibiting Cdk1/cyclinB and Cdk5/p25, also with an approximate IC50 of 20 μM. This compound is notable for its ability to reduce spindle checkpoint-signaling proteins, such as Mad2 and BubR1, at the kinetochores of chromosomes, making it a valuable tool in cancer research and studies on mitotic regulation. -
Aurora Kinase Inhibitor
AT9283 hydrochloride is a multi-targeted kinase inhibitor that primarily targets Aurora A and Aurora B kinases, which play critical roles in cell proliferation and survival. Its inhibitory effects extend to additional kinases such as JAK2 and Abl (T315I), enhancing its potential utility in cancer research. AT9283 hydrochloride has demonstrated significant anti-tumor activity, making it a valuable tool for investigating therapeutic strategies in various malignancies. -
Aurora Kinase Inhibitor
VE-465 is a potent Aurora kinase inhibitor that promotes apoptosis in cancer cells. Its anticancer activities have been demonstrated across various tumor models, making it a valuable tool for cancer research. This compound's ability to selectively target Aurora kinases positions it as a significant candidate for investigations into tumor progression and treatment strategies. -
Aurora Kinase Inhibitor
Binucleine 2 is an ATP-competitive inhibitor targeting Drosophila Aurora B kinase, displaying an inhibition constant (Ki) of 0.36 μM. This compound exhibits isoform specificity, effectively inhibiting Drosophila Aurora B in a dose-dependent manner while exhibiting minimal activity against human and Xenopus laevis Aurora B kinases at concentrations up to 100 μM. Binucleine 2 is valuable for studying mitotic processes, as it induces defects in mitosis and cytokinesis in Drosophila Kc167 cells and disrupts contractile ring assembly in Drosophila S2 cells at a concentration of 40 μM, highlighting the critical role of Aurora B kinase in cell division. -
Aurora A Inhibitor
Aurora A inhibitor 4 (compound C9) is a selective inhibitor of Aurora A kinase, demonstrating a GI50 of 4.26 μM in DU 145 cells and 7.08 μM in HT-29 cells. This compound exhibits significant anti-proliferative effects, making it valuable for research focused on cell cycle regulation and cancer therapeutics. Its ability to inhibit Aurora A activity can be applied in studies investigating mitotic dysregulation in various tumor types. -
Aurora Kinase Control
Win 47338 is a control compound targeting Aurora kinases (AurA/AurB) and the mitotic kinase monopolar spindle 1 (MPS1). It serves as a crucial reference for studies involving mitotic regulation and cellular division processes. With a Ki value greater than 100 μM, it provides a baseline for evaluating the potency of other kinase inhibitors in research applications focused on cell cycle dynamics and cancer therapeutics. -
Aurora Kinase Inhibitor
Aurora B inhibitor 1 is a selective inhibitor of the Aurora B kinase, exhibiting a Ki value of <0.010 µM. This compound plays a crucial role in manipulating cell division and is instrumental in research surrounding cancer biology and therapeutic development. Its ability to inhibit Aurora B activity makes it a valuable tool for studies focused on mitotic regulation and chromosomal instability. -
BRD4 PROTAC Degrader
PROTAC BRD4 Degrader-21 is a targeted PROTAC that degrades the BRD4 protein through the induction of ubiquitination, achieving an IC50 of 59 nM. This compound effectively leads to BRD4 degradation via the proteasome pathway, and demonstrates moderate affinity for recombinant HSP90α with an IC50 range of 100-1000 nM. In preclinical studies, PROTAC BRD4 Degrader-21 has been shown to induce apoptosis in cancer cells and inhibit tumor growth in xenograft mouse models, making it a valuable tool for research into acute myeloid leukemia and diffuse large B-cell lymphoma. -
JAK/STAT Inhibitor
Phenylpyropene C is a JAK/STAT pathway inhibitor known to effectively inhibit interferon-gamma (IFN-γ) mediated expression of reporter genes, with an IC50 range of 5.4 to 10.8 μM. Additionally, it serves as an inhibitor of acyl-CoA, exhibiting an IC50 of 16.0 μM. This compound is valuable for research applications focusing on the modulation of immune responses and the exploration of cytokine signaling pathways. -
HDAC Inhibitor
2-Propylpent-4-ynoic acid, a histone deacetylase (HDAC) inhibitor, exhibits an IC50 of 0.5 mM against human HDAC. This compound induces P-glycoprotein function and has been associated with teratogenicity, fetal growth inhibition, and neurotoxicity. Notably, the S-enantiomer demonstrates more significant teratogenic effects compared to its R-enantiomer and other analogs. 2-Propylpent-4-ynoic acid is relevant in research focused on the mechanisms underlying colon cancer and neural tube defects, including exencephaly. -
Sirtuin-modulating Compound
Sirtuin Modulator 9 is a sirtuin-modulating compound that enhances cellular lifespan and promotes mitochondrial activity. This compound has potential applications in research related to aging, inflammation, and cancer. Investigators may utilize Sirtuin Modulator 9 to explore its therapeutic effects in age-related disorders and diseases associated with compromised mitochondrial function. -
Endogenous Metabolite
5-Hydroxymethylcytosine (5hmC) is an oxidized derivative of 5-methylcytosine found within mammalian DNA. Generated through an enzymatic process involving the Ten-eleven translocation (TET) enzymes TET1, TET2, and TET3, 5hmC serves as an important marker for investigating the dynamic processes of DNA demethylation and gene transcription. Its presence is particularly significant in the study of conditions such as non-small cell lung cancer, neurodegenerative diseases like Alzheimer’s and Parkinson’s, and various hematological malignancies, including acute myeloid leukemia and myelodysplastic syndromes. -
AMPK/SIRT3/PGC-1α Modulator
MitoPBN is an AMPK/SIRT3/PGC-1α modulator that enhances mitochondrial function by acting as a reactive oxygen species scavenger. This compound promotes mitochondrial biogenesis through increased AMPK phosphorylation, restoration of SIRT3 expression, and upregulation of PGC-1α. MitoPBN is effective in regulating glucose metabolism, as it decreases blood glucose levels by inhibiting hepatic gluconeogenesis and enhancing glucose uptake, while also improving ATP production and maintaining mitochondrial membrane potential. Additionally, it can reduce apoptosis and enhance sperm motility and membrane integrity, making it a valuable reagent for research related to diabetes and metabolic disorders. -
Stable Isotope
SAH-13C10 is a stable isotope labeled derivative of S-Adenosylhomocysteine (SAH). As an important amino acid derivative, SAH plays a crucial role as a modulator in various metabolic pathways and serves as an intermediate in the synthesis of cysteine and adenosine. It functions as an inhibitor of the METTL3-METTL14 heterodimer complex, demonstrating an IC50 of 0.9 μM, making it valuable for research in epitranscriptomics and cellular metabolism studies. -
WDR5 Ligand
Dimethyl-F-OICR-9429-COOH is a potent ligand for the WD40 repeat domain protein 5 (WDR5), functioning as a critical component in the synthesis of proteolysis-targeting chimeras (PROTACs). Its specific binding to WDR5 facilitates the targeted degradation of associated proteins, making it a valuable tool in chemical biology research. This compound is instrumental in the exploration of protein interactions and the development of innovative therapeutic strategies. -
SMARCA2 Ligand
SMARCA2 ligand-8 acts as a selective ligand for the target protein SMARCA2, facilitating the synthesis of the PROTAC SMARCA2/4-degrader-35. This compound is instrumental in research applications focused on targeted protein degradation, offering insights into the modulation of gene regulation and chromatin remodeling. Its utility in developing innovative degraders highlights its significance in the study of cancer biology and potential therapeutic interventions. -
JAK3/TEC Family Kinase Inhibitor
Plodicitinib is an inhibitor of Janus tyrosine kinase 3 (JAK3) and TEC family kinases. This compound exhibits significant anti-inflammatory activity, making it valuable for research in inflammation-related disorders. It is applicable in studies exploring the modulation of immune responses and potential therapeutic interventions in autoimmune diseases. -
BRD4 BD2 Inhibitor
BRD4-BD1/2-IN-3 is a selective inhibitor of the BRD4 bromodomain 2 (BD2), exhibiting an IC50 of 0.41 nM for BRD4 BD2 compared to BRD4 BD1. This compound effectively inhibits LPS-induced expression of IL-6, demonstrating significant anti-inflammatory properties through modulation of the TNF and NF-κB signaling pathways. BRD4-BD1/2-IN-3 is valuable for research focused on inflammatory diseases. -
PARP-1 Inhibitor
L-2286 is a potent orally active inhibitor of PARP-1. This compound demonstrates significant biological activity by alleviating carotid artery remodeling, reducing oxidative stress and inflammation in spontaneously hypertensive rats, while also providing neuroprotective effects in the dorsal hippocampus. L-2286 is applicable in research focused on hypertension and its associated vascular and neurological complications. -
BRD4 PROTAC
Lenalidomide-CO-C7-NH2 is a CRBN-dependent intermediate designed as a BRD4-targeting PROTAC degrader. This compound facilitates the selective degradation of the oncoprotein BRD4, leading to decreased cancer cell proliferation, cell cycle arrest, and enhanced apoptosis. It demonstrates noteworthy anti-tumor efficacy in xenograft models, making Lenalidomide-CO-C7-NH2 a valuable tool for investigating mechanisms in acute myeloid leukemia research. -
Fluorescent Probe
Citrulline-specific probe-biotin is a biotinylated fluorescent probe designed for the detection of citrulline, a hydrolysis product of arginine produced by the enzyme protein arginine deiminase (PAD). Increased PAD activity is implicated in various pathological conditions, including autoimmune diseases and inflammatory disorders. This probe facilitates the identification of diseases characterized by elevated citrulline levels and is particularly useful in animal models of ulcerative colitis for studying disease mechanisms and potential therapeutic interventions.
