Epigenetics
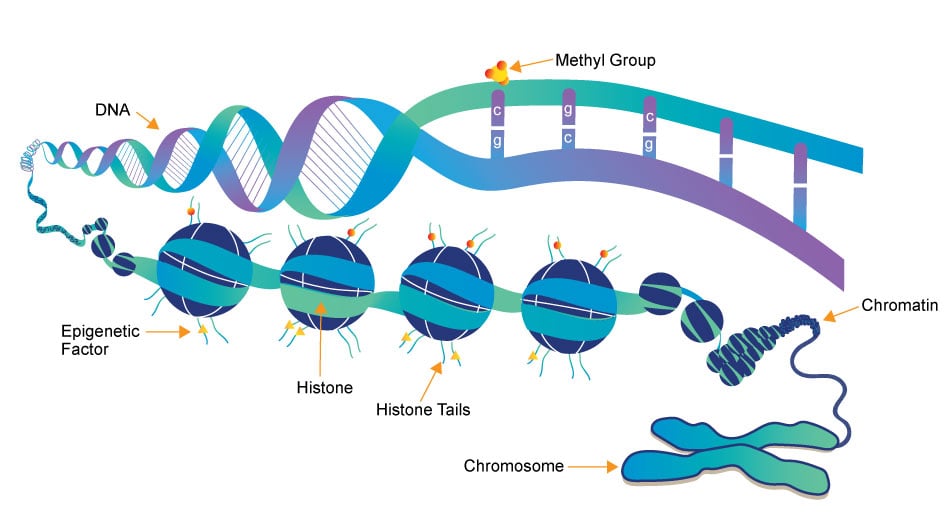
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
TOPOI/PARP Dual Inhibitor
TOPOI/PARP-1-IN-1 is a dual inhibitor targeting topoisomerase I and PARP-1, demonstrating an IC50 value of 0.09 μM for PARP-1. This compound exhibits significant anti-proliferative and anti-migratory effects on cancer cells, leading to G0/G1 phase cell cycle arrest and apoptosis. In preclinical studies, TOPOI/PARP-1-IN-1 achieved a tumor growth inhibition rate of 75.4% in mice, highlighting its potential for cancer therapy research applications. -
LSD1/KDM1A Inhibitor
TPC-144 is an inhibitor of LSD1/KDM1A, targeting the demethylation process by reducing DNMT1 protein levels, resulting in decreased methylation of LINE-1 elements. This compound has shown promising synergistic effects with Decitabine, enhancing DNA demethylation, which promotes differentiation and apoptosis in leukemia cells. Furthermore, TPC-144 has demonstrated anti-tumor activity in acute myeloid leukemia (AML) models, making it a valuable tool for AML research and therapeutic studies. -
SIRT6 Inhibitor
SIRT6-IN-4 is a selective inhibitor of SIRT6, demonstrating an IC50 of 5.68 μM. This compound effectively inhibits the proliferation of MCF-7 cells with an IC50 of 8.30 μM, leading to cell cycle arrest at the G2/M phase. Additionally, SIRT6-IN-4 reduces cell migration and invasion while inducing apoptosis. Its antitumor efficacy has been confirmed in mouse models, making it a valuable tool for cancer research and therapeutic development. -
JAK2/Bcr-Abl/FLT3 Inhibitor
LS-104 is a non-ATP-competitive inhibitor targeting JAK2, Bcr-Abl, and FLT3. It effectively induces apoptosis in JAK2V617F-positive cells while inhibiting JAK2 autophosphorylation and downstream signaling pathways. Additionally, LS-104 demonstrates significant cytotoxic effects and inhibits the proliferation of FLT3-expressing leukemic cells. This hydroxystyryl-acrylonitrile compound holds potential for research into myeloproliferative disorders and refractory or relapsed hematologic malignancies. -
SIRT1 Agonist
BF-175 is a selective agonist of SIRT1, a protein involved in cellular regulation and energy metabolism. It enhances the activation of PGC1-α, promoting autophagy and apoptosis, while also inhibiting the activity of SREBP. BF-175 demonstrates protective effects against high glucose-induced mitochondrial damage and shows potential in attenuating the progression of diabetic kidney disease. Additionally, this compound has been investigated for its inhibitory effects on endometrial carcinoma, making it a valuable tool for research in metabolic and cancer studies. -
PARP/NAMPT Inhibitor
PARP1/NAMPT-IN-2 is a potent dual inhibitor of PARP1 and NAMPT, exhibiting IC50 values of 0.8 nM and 18 nM, respectively. This compound effectively inhibits cell proliferation and migration, while inducing apoptosis in breast cancer cells. PARP1/NAMPT-IN-2 is particularly relevant for investigating therapeutic strategies in triple-negative breast cancer research. -
JAK3 Inhibitor
PRN-371 is a potent and selective inhibitor of JAK3. By disrupting the JAK3-STAT signaling pathway, PRN-371 effectively suppresses the proliferation of natural killer and T-cell lymphoma cells, inducing apoptosis in these malignancies. This compound demonstrates significant antitumor activity and is applicable in the research of various cancer types, particularly hematological malignancies. -
JAK2 Inhibitor
JAK2-IN-14 is a highly selective JAK2 inhibitor with an IC50 of 2 nM, demonstrating significant selectivity with 89.5-fold over JAK1, 80.5-fold over JAK3, and 51-fold over TYK2. This compound effectively inhibits the STAT5 signaling pathway, leading to tumor cell cycle arrest and apoptosis. JAK2-IN-14 is a valuable tool for investigating myeloproliferative neoplasms (MPNs) and their underlying mechanisms. -
MLL1 Inhibitor
MM-401 is a potent inhibitor of MLL1, a methyltransferase that targets H3K4. By interfering with the MLL1-WDR5 interaction, MM-401 effectively inhibits MLL1 activity with an IC50 of 0.32 μM. This compound has been shown to induce cell cycle arrest, promote apoptosis, and facilitate differentiation. MM-401 is particularly relevant for research focused on MLL leukemia. -
PARP1/2/CDK12 Inhibitor
PARP-1/2-IN-2 is a potent inhibitor of PARP1, PARP2, and CDK12, exhibiting IC50 values of 34 nM, 30 nM, and 285 nM, respectively. This compound disrupts DNA damage repair mechanisms, leading to induced cell cycle arrest and apoptosis. Notable for its efficacy in targeted therapy, PARP-1/2-IN-2 effectively inhibits the growth of triple-negative breast cancer (TNBC) cells and demonstrates significant antitumor activity in TNBC xenograft models. This makes it a valuable tool for research in cancer biology and therapeutic development. -
PIM1 Inhibitor
PIM1-IN-3 is a selective inhibitor of the PIM1 kinase, known for its role in promoting cell survival and proliferation. This compound effectively induces apoptosis in Colo320 cells, demonstrating its potential as a therapeutic agent in cancer research. PIM1-IN-3 serves as a valuable tool for studying PIM1-related signaling pathways and exploring targeted cancer treatments. -
Aurora-A kinase Inhibitor
LY3295668 erbumine is a potent and selective inhibitor of Aurora-A kinase, exhibiting a Ki value of 0.8 nM for AurA while demonstrating significantly lower binding affinity for AurB at 1038 nM. This compound effectively inhibits the autophosphorylation of Aurora-A, leading to mitotic arrest and apoptosis without promoting polyploidy associated with AurB inhibition. LY3295668 erbumine is valuable for research into small cell lung cancer and other conditions involving dysregulation of Aurora kinases. -
HDAC/PSMD14 Inhibitor
HDAC/PSMD14-IN-1 is a dual-target inhibitor of HDAC1 and PSMD14, exhibiting IC50 values of 238.7 nM and 141.2 nM, respectively. This compound demonstrates significant cytotoxicity against esophageal squamous cell carcinoma (ESCC) cell lines, with IC50 values ranging from 30 to 250 nM. In addition to its ability to induce apoptosis, HDAC/PSMD14-IN-1 effectively reverses epithelial-mesenchymal transition (EMT) and shows promising anti-tumor activity in KYSE30 mouse xenograft models. It is a valuable tool for advancing research in esophageal cancer. -
HDAC Inhibitor
1-Alaninechlamydocin is a cyclic tetrapeptide that functions as a potent histone deacetylase (HDAC) inhibitor with an IC50 of 6.4 nM. This compound effectively induces G2/M cell cycle arrest and promotes apoptosis in MIA PaCa-2 cells, making it a valuable tool for cancer research. Its activity in modulating epigenetic regulation highlights its potential applications in therapeutic development and studies of cellular differentiation and survival. -
HDAC Class I Inhibitor
HDAC-IN-27 is a selective inhibitor of Class I histone deacetylases (HDAC1-3) with an IC50 range of 0.43 to 3.01 nM. It demonstrates significant anti-proliferative and pro-apoptotic effects against acute myeloid leukemia (AML) cell lines by promoting histone acetylation, specifically AcHH3 and AcHH4. This compound is valuable for research into the mechanisms of AML and potential therapeutic applications in histone modification regulation. -
Pim-1 Inhibitor
Pim-1 kinase inhibitor 1 is a selective inhibitor of Pim-1 kinase, demonstrating an IC50 value of 0.11 μM. This compound exhibits significant anticancer activity across various cancer cell lines by promoting cellular apoptosis. Pim-1 kinase inhibitor 1 is a valuable tool for research applications focused on cancer biology and therapeutic development. -
HDAC6 Inhibitor
Daphnegiravone D is an inhibitor of HDAC6, targeting histone deacetylation to modulate gene expression. This compound demonstrates significant anti-hepatocellular carcinoma activity by inducing apoptosis and selectively inhibiting the proliferation of liver cancer cells. Its mechanism involves the p38 and JNK MAPK signaling pathways, making it a valuable tool for research in cancer therapeutics and cellular signaling. -
Aurora B Inhibitor
HOI-07 is a selective inhibitor of Aurora B kinase that interferes with the phosphorylation of histone H3 at Ser10 in lung cancer cells. This compound induces cell-cycle arrest and apoptosis, demonstrating significant antitumor activity. HOI-07 effectively suppresses tumor growth in xenograft models, including A549, 143B, and KHOS, making it a valuable tool for cancer research and therapeutic investigation. -
HDAC1/6 Inhibitor
HDAC1/6-IN-1 is a potent inhibitor targeting HDAC1 and HDAC6, exhibiting IC50 values of 1.3 nM and 13 nM, respectively. This compound effectively inhibits the methylation and deacetylation of H3K9, leading to significant biological activity, including the induction of apoptosis in cancer cells, G0/G1 cell cycle arrest, and the inhibition of cell migration and invasion. It serves as a valuable tool in cancer research and the study of epigenetic regulation. -
Dual TOP1/PARP1 Inhibitor
DiPT-4 is a dual inhibitor of topoisomerase I (TOP1) and poly (ADP-ribose) polymerase 1 (PARP1). This compound induces substantial DNA double-strand breaks, leading to cell cycle arrest and apoptosis in various cancer cell lines. DiPT-4 is particularly valuable for research focused on overcoming mechanisms of cancer drug resistance. -
PARP Inhibitor
Schisandronic acid is a potent PARP inhibitor derived from the triterpenoid compound found in Schisandra chinensis. This compound exhibits significant cytotoxicity against human breast cancer cells, particularly MCF-7, with an IC50 value of 8.06 μM. Schisandronic acid effectively induces apoptosis through the upregulation of active caspase-3 and cleavage of PARP, while also reducing reactive oxygen species generation, thereby demonstrating notable antioxidant properties. Its mechanisms of action make Schisandronic acid a valuable tool for cancer research and therapeutic investigations. -
CBP Bromodomain Inhibitor
Ischemin is a selective inhibitor of the CBP bromodomain, effectively disrupting the interaction between p53 and CBP, consequently reducing transcriptional activity. With an IC50 value of 5 µM, Ischemin demonstrates the ability to inhibit p53-induced p21 activation. Additionally, it has been shown to protect against apoptosis in ischemic cardiomyocytes. This reagent is valuable for investigating mechanisms involved in cardiovascular diseases, particularly myocardial ischemia. -
SIRT2 Inhibitor
SIRT2-IN-18 is a selective SIRT2 inhibitor, exhibiting IC50 values of 5.3 μM for SmSIRT2 and 12.3 μM for hSIRT2. This compound demonstrates significant antischistosomal effects against both Liberian and Puerto Rican strains of Schistosoma mansoni, effectively reducing schistosomula viability, adult worm pairing, and egg production while maintaining low cytotoxicity in mammalian cells. SIRT2-IN-18 also promotes histone H3 hyperacetylation and triggers cytochrome c-mediated apoptosis, making it a valuable tool for research into both parasitic infections and the modulation of acetylation pathways. -
JAK3 Inhibitor
NSC114792 is a selective inhibitor of Janus kinase 3 (JAK3), which plays a critical role in immune signaling pathways. This compound effectively induces apoptosis in target cells and significantly reduces the protein expression of phosphorylated JAK3 and phosphorylated STAT5. NSC114792 is primarily utilized in research focused on immune responses and related signaling cascades. -
HDAC Inhibitor
HDAC-IN-39 is a potent inhibitor of histone deacetylases (HDACs), exhibiting IC50 values of 1.07 μM for HDAC1, 1.47 μM for HDAC2, and 2.27 μM for HDAC3. This compound also significantly disrupts microtubule polymerization and induces cell cycle arrest at the G2/M phase, highlighting its potential for modulating cell cycle dynamics. Furthermore, HDAC-IN-39 demonstrates promising anticancer activity, particularly against resistant cancer cell lines, making it a valuable tool for cancer research and therapeutic exploration. -
Topoisomerase/HDAC Inhibitor
Top/HDAC-IN-3 is an orally active dual inhibitor targeting topoisomerase and histone deacetylase (HDAC). This compound enhances intracellular levels of reactive oxygen species (ROS), leading to DNA damage and subsequently inhibiting cancer cell colony formation and migration. Additionally, Top/HDAC-IN-3 induces apoptosis and cell cycle arrest in cancer cells. In non-small cell lung cancer (NSCLC) models, it demonstrates significant antitumor activity, achieving a tumor growth inhibition (TGI) of 77.5% at a dosage of 100 mg/kg. -
PARP1/BRD4 Inhibitor
PARP1/BRD4-IN-1 is a selective inhibitor targeting both PARP1 and BRD4, demonstrating IC50 values of 49 nM and 202 nM, respectively. This compound effectively represses the expression and activity of these proteins, leading to synergistic inhibition of malignant pancreatic cancer cell growth. PARP1/BRD4-IN-1 is a valuable tool for exploring therapeutic strategies in cancer research, particularly in the context of PARP and BRD4 signaling pathways. -
HDAC6 Inhibitor
TNI-97 is a highly selective and orally active inhibitor of histone deacetylase 6 (HDAC6), exhibiting an IC50 of 0.2 nM. This compound effectively suppresses the growth and clonogenicity of triple-negative breast cancer (TNBC) cells, specifically MDA-MB-453. TNI-97 induces PANoptosis, encompassing apoptosis, necroptosis, and pyroptosis in these cells. Additionally, TNI-97 demonstrates significant antitumor activity in mouse models, including xenografts and allografts of TNBC, making it a valuable tool for research focused on triple-negative breast cancer. -
HDAC6 Inhibitor
PTG-0861 is a selective inhibitor of histone deacetylase 6 (HDAC6) with an IC50 value of 5.92 nM. This compound effectively induces apoptosis, making it a valuable tool for research in acute myeloid leukemia, multiple myeloma, and other hematological malignancies. Its specificity towards HDAC6 positions it as a promising candidate for studies aimed at understanding epigenetic regulation in cancer. -
Pan-Pim kinase Inhibitor
VS-II-173 is a potent pan-Pim kinase inhibitor, exhibiting IC50 values of 0.07 μM for Pim1 and 0.02 μM for Pim3, with a residual activity of 46% at 1 μM for Pim2. This compound selectively targets acute myeloid leukemia (AML) cells, demonstrating significant inhibition of key phosphorylation events, including Stat5 (Y694) and MDM2 (S166), which disrupts pro-survival signaling pathways and promotes apoptosis. VS-II-173 shows enhanced anti-AML efficacy when used in combination with Daunorubicin and is especially relevant for research involving AML characterized by FLT3-ITD and NPM1 mutations. Its minimal toxicity to non-malignant cells makes it a valuable tool in cancer research. -
HDAC/ Topo II α Inhibitor
KT32 is a potent dual inhibitor targeting histone deacetylases (HDAC) and topoisomerase II alpha (Topo II α). This compound promotes cell death through the activation of apoptotic pathways, making it valuable for research in cancer biology and therapeutic studies. KT32's ability to modulate chromatin structure and DNA topology renders it an essential tool for exploring the mechanisms of tumor progression and treatment resistance. -
HDAC Inhibitor
(E/Z)-Dacinostat is a potent histone deacetylase (HDAC) inhibitor that plays a critical role in inducing apoptosis in cancer cells, particularly leukemia. By promoting the generation of reactive oxygen species (ROS) and instigating DNA damage, (E/Z)-Dacinostat enhances the cytotoxic efficacy of fludarabine against leukemia cells. Its mechanism involves modulation of DNA repair pathways and intracellular signaling, making it a valuable tool for cancer research and therapeutic investigations. -
Aurora Kinase Inhibitor
Aurora Kinase Inhibitor-14 is a highly selective inhibitor of Aurora kinases, demonstrating IC50 values of 0.5 nM for Aurora A and 1.2 nM for Aurora B. This compound binds to the ATP-binding site of these kinases, effectively disrupting chromosome segregation during mitosis and promoting apoptosis in tumor cells. Aurora Kinase Inhibitor-14 is a valuable tool for investigating the therapeutic potential in various solid tumors and hematological malignancies, including non-small cell lung cancer, breast cancer, and acute myeloid leukemia. -
HDAC Inhibitor
SK-7041 is a histone deacetylase (HDAC) inhibitor with an IC50 value of 172 nM. This compound promotes hyperacetylation of histones H3 and H4, leading to the inhibition of tumor cell growth both in vitro and in vivo. Additionally, SK-7041 induces apoptosis and causes cell cycle arrest at the G1 phase, making it a valuable tool for cancer research and therapeutic exploration. -
HDAC8 Inhibitor
HDAC8-IN-3 is a potent inhibitor of Histone Deacetylase 8 (HDAC8), exhibiting an IC50 value of 9.3 μM. This compound induces thermal stabilization and demonstrates cytotoxic effects, leading to apoptosis in leukemic cell lines. HDAC8-IN-3 is valuable for research applications focused on cancer metabolism, epigenetic regulation, and therapeutic development for hematological malignancies. -
Hsp90/HDAC6 Inhibitor
HDAC6/HSP90-IN-2 is a dual inhibitor targeting both HDAC6 and Hsp90, exhibiting IC50 values of 105.7 nM and 61 nM, respectively. This compound demonstrates significant potential in cancer research, enabling the study of mechanisms involved in tumorigenesis and the development of novel therapeutic strategies. Its ability to modulate key cellular pathways associated with cancer progression makes it a valuable tool for investigating the role of HDAC6 and Hsp90 in various malignancies. -
DNMT Inhibitor
DNMT1-IN-5 is a selective inhibitor targeting DNA methyltransferases DNMT1 and DNMT3A, with IC50 values of 2.42 μM and 14.4 μM, respectively. This compound demonstrates significant antiproliferative activity across various cancer cell lines, exhibiting IC50 values ranging from 0.19 to 2.37 μM in TMD-8, DOHH2, MOLM-13, THP-1, RPIM-8226, and HCT116. In addition to inducing cell cycle arrest at the G2/M phase, DNMT1-IN-5 promotes apoptosis in TMD-8 and DOHH2 cells and has shown antitumor efficacy in xenograft mouse models. -
Wee1/HDAC Inhibitor
Wee1/HDAC-IN-1 is a dual inhibitor targeting Wee1 and histone deacetylases (HDACs). It demonstrates potent activity with an IC50 of 1.2 nM for Wee1 and varying IC50 values of 196 nM for HDAC1, 156 nM for HDAC3, and 55 nM for HDAC6. This compound displays significant antiproliferative effects in MV4-11 cells, with an IC50 of 0.076 μM, by disrupting DNA damage repair mechanisms and promoting apoptosis. Wee1/HDAC-IN-1 is suited for research on acute myeloid leukemia (AML). -
HDAC/DNMT Inhibitor
J208 is a dual inhibitor targeting histone deacetylase (HDAC) and DNA methyltransferase (DNMT). This compound effectively inhibits the proliferation of cancer cells and reduces the migration and invasion of triple-negative breast cancer (TNBC) cells. J208 also induces apoptosis and halts the cell cycle at the G0/G1 phase, while activating innate immune signaling pathways by promoting the expression of endogenous retroviruses (ERVs) in TNBC. It serves as a valuable tool for investigating epigenetic regulation and cancer therapy. -
ARP-1/HDAC-1 Inhibitor
DLC-50 is a dual inhibitor of PARP-1 and HDAC-1, exhibiting IC50 values of 1.2 nM and 31 nM, respectively. This compound effectively inhibits the proliferation of various breast cancer cell lines, including MDA-MB-436, MDA-MB-231, and MCF-7, with IC50 values of 0.3, 2.7, and 2.41 μM. Additionally, DLC-50 induces apoptosis specifically in MDA-MB-231 cells and causes cell cycle arrest at the G2 phase, making it a valuable tool for cancer research and therapeutic development. -
CDK9/HDAC Dual Inhibitor
CDK9/HDAC1/HDAC3-IN-1 is a dual inhibitor targeting CDK9 and HDACs. With IC50 values of 0.17 μM for CDK9, 1.73 μM for HDAC1, and 1.11 μM for HDAC3, this compound effectively disrupts the activity of these proteins. It induces cancer cell apoptosis and causes cell cycle arrest at the G2/M phase. Additionally, CDK9/HDAC1/HDAC3-IN-1 exhibits broad-spectrum anti-cancer effects, demonstrating efficacy against various malignancies, including breast, cervical, and liver cancers, as evidenced in murine TNBC MDA-MB-231 xenograft models. -
HDAC1/2 Inhibitor
ZWZH-21 is a selective inhibitor of histone deacetylases HDAC1 and HDAC2, demonstrating IC50 values of 34 nM and 41 nM, respectively. This dual-action compound exhibits potent anti-proliferative effects on colorectal cancer cell lines HCT116 and SW480, with IC50 values of 0.524 μM and 1.063 μM, respectively. Additionally, ZWZH-21 effectively inhibits cell migration and prompts apoptosis in multiple colorectal cancer models, making it a valuable tool for cancer research, particularly in the study of colorectal cancer. -
BRD4 Inhibitor
BET-IN-20 is a selective BRD4 bromodomain inhibitor with an IC50 of 1.9 nM. This compound demonstrates significant anticancer activity by inducing apoptosis in acute myeloid leukemia (AML) cells and effectively arresting the cell cycle in the G0/G1 phase. Additionally, BET-IN-20 inhibits c-Myc and CDK6, while enhancing PARP cleavage, making it a valuable tool for cancer research and therapeutic development. -
BRD4 PROTAC Degrader
NEP162 is a potent BRD4 PROTAC degrader, demonstrating DC50 values of 1.2 and 1.6 μM in SW480 and U2OS cell lines, respectively. It exhibits significant antiproliferative activity, effectively inhibiting tumor growth and promoting apoptosis in various cancer models. NEP162 is particularly relevant for research applications in osteosarcoma, colorectal cancer, and non-small cell lung cancer. -
Aurora A Inhibitor
Aurora A Inhibitor 2 is a potent inhibitor of Aurora A kinase, exhibiting an IC50 value of 21.94 nM. This compound has been shown to induce caspase-dependent apoptosis in MDA-MB-231 cells, highlighting its potential as a therapeutic agent in cancer research. It is useful for studies investigating the role of Aurora A in cell cycle regulation and apoptosis. -
HDAC1/HDAC2 Inhibitor
ST13 is a selective inhibitor of HDAC1 and HDAC2, exhibiting IC50 values of 23 nM and 49 nM, respectively. It offers weak inhibition of HDAC3 and HDAC6, with IC50 values of 4.30 μM and >10 μM, respectively. The binding mechanism of ST13 involves an initial rapid formation of a collision complex followed by a slow conversion to a stable complex. This compound has demonstrated the ability to induce apoptosis in cancer cells and is useful for research on melanoma and triple-negative breast cancer. -
Topo II/ HDAC Inhibitor
Topo II/HDAC-IN-1 is a potent dual inhibitor targeting Topoisomerase II (Topo II) and histone deacetylases (HDACs). This compound is known to induce apoptosis in cancer cells, making it a valuable tool for research in cancer biology and therapeutic development. Its ability to simultaneously inhibit these targets can provide insights into novel cancer treatment strategies. -
PDE5/HDAC Inhibitor
PDE5/HDAC-IN-1 is a dual inhibitor of phosphodiesterase 5 (PDE5) and histone deacetylases (HDAC) with IC50 values of 46.3 nM and 14.5 nM, respectively. This compound has demonstrated the capability to induce cell apoptosis and exhibits significant anticancer activities. PDE5/HDAC-IN-1 is a valuable tool for research in cancer therapeutics and epigenetic modulation. -
HDAC3 Inhibitor
HDAC3-IN-6 is a selective inhibitor of histone deacetylase 3 (HDAC3) with an IC50 of 53 nM. This compound effectively induces the expression of PD-L1 in a dose-dependent manner, promoting apoptosis and elevating reactive oxygen species (ROS) production. HDAC3-IN-6 demonstrates significant antitumor efficacy, particularly in colorectal cancer models, making it a valuable tool for research into cancer therapy and immunomodulation. -
HDAC1-3 PROTAC Degrader
JPS004 is a targeted proteolysis targeting chimera (PROTAC) that degrades histone deacetylases HDAC1-3. By inducing the degradation of these enzymes, JPS004 facilitates histone acetylation, which can promote apoptosis in cancer cells. This compound is valuable for research into cancer biology and therapeutic strategies aimed at modulating epigenetic modifications.
