Epigenetics
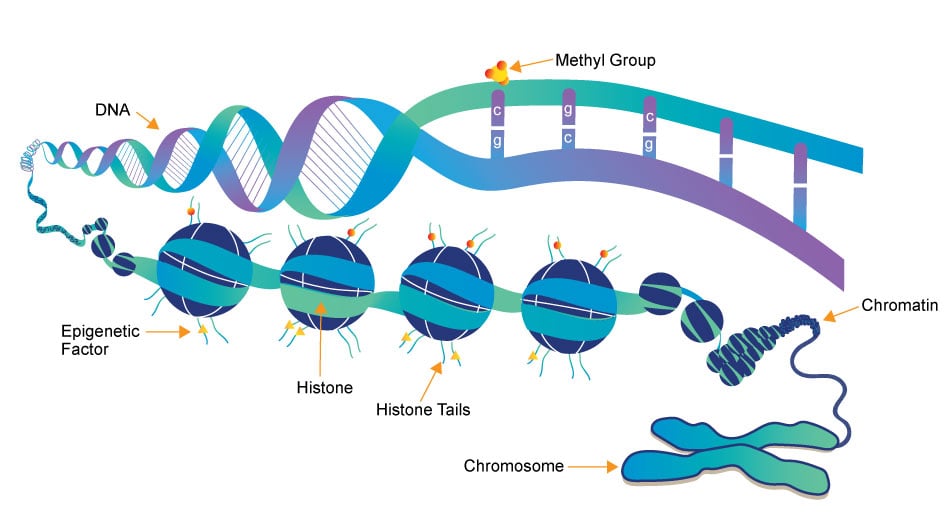
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
LSD1 Inhibitor
cis-4-Br-2,5-F2-PCPA is a selective inhibitor of lysine-specific demethylase 1 (LSD1), demonstrating a Ki value of 94 nM for LSD1 compared to 8.4 μM for LSD2. This compound effectively inhibits LSD1 activity, resulting in reduced proliferation of cancer stem cells by increasing the levels of dimethylated histone H3 at lysine 4 (H3K4) in CCRF-CEM cells. Its selective action makes cis-4-Br-2,5-F2-PCPA a valuable tool for research on epigenetic regulation and cancer biology. -
KDM5 Demethylases Inhibitor
KDM5-C49 is a potent and selective inhibitor of KDM5 demethylases, exhibiting IC50 values of 40 nM, 160 nM, and 100 nM for KDM5A, KDM5B, and KDM5C, respectively. This compound plays a critical role in epigenetic research, particularly in the study of cancer biology. It provides valuable insights into the mechanisms of gene regulation and the potential therapeutic strategies targeting KDM5 demethylases in cancer treatment. -
LSD1 Inhibitor
LSD1-IN-48 is a selective inhibitor of lysine-specific demethylase 1 (LSD1), featuring a tranylcypromine-pyrimidine structure with a human IC50 of 7.87 nM. This compound enhances histone methylation levels of H3K4me1 and H3K4me2, promoting apoptosis in cancer cells. Additionally, LSD1-IN-48 upregulates CD86 and downregulates SOX2 and CD44, leading to reduced cell proliferation. It is particularly useful for research into acute myeloid leukemia. -
LSD1 Inhibitor
LSD1-IN-35 is a selective inhibitor of lysine-specific demethylase 1 (LSD1) with an IC50 of 108 nM. By inhibiting the demethylation of H3K4me1/2, LSD1-IN-35 acts as an immunomodulator, enhancing the response of gastric cancer cells to T-cell-mediated cytotoxicity. This compound decreases PD-L1 expression, thereby mitigating the PD-1/PD-L1 interaction, which is critical in tumor immune evasion. Its potential applications include cancer research and the study of immune regulation in tumor microenvironments. -
LSD1 Inhibitor
LSD1-IN-18 is a selective, non-covalent inhibitor of LSD1, exhibiting a Ki of 0.156 μM and a KD of 0.075 μM. It demonstrates significant antiproliferative effects in THP-1 leukemia cells and MDA-MB-231 breast cancer cells, with calculated IC50 values of 0.16 μM and 0.21 μM, respectively, after 72 hours of treatment. This compound provides a valuable tool for investigating the role of LSD1 in cancer biology and therapeutic applications. -
KDM5A/5B Histone Lysine Demethylase Inhibitor
N19-0881 is a selective inhibitor of KDM5A and KDM5B histone lysine demethylases, exhibiting potent activity with IC50 values of 0.013 μM and 0.002 μM, respectively. This compound demonstrates significant potential in the study of epigenetically dysregulated tumors, particularly in applications related to breast cancer research. Its oral bioactivity makes it an effective tool for investigating the role of histone modification in cancer biology and therapeutic development. -
LSD1 Inhibitor
Bizine di(hydrochloride) is a potent and selective inhibitor of the lysine-specific demethylase 1 (LSD1) enzyme, exhibiting a Ki value of 59 nM. This compound effectively modulates bulk histone methylation in cancer cells, making it valuable for epigenetic studies related to cancer biology. Additionally, Bizine di(hydrochloride) demonstrates neuroprotective effects, supporting its potential applications in neurological research. -
LSD1 Inhibitor
3β-Acetoxyl-atractylenolide I is a potent inhibitor of lysine-specific demethylase 1 (LSD1), demonstrating an IC50 of 57 μM. This compound has been shown to effectively block tumor growth, metastasis, and invasion across various cancer types. It is utilized in research focusing on prostate cancer, breast cancer, neuroblastoma, gastric cancer, colon cancer, bladder cancer, esophageal cancer, acute myeloid leukemia, and retinoblastoma. -
JMJD3/UTX Inhibitor
GSK-J4 hydrochloride is a potent dual inhibitor of the demethylases JMJD3 (KDM6B) and UTX (KDM6A), with IC50 values of 8.6 μM and 6.6 μM, respectively. This compound demonstrates significant inhibition of LPS-induced TNF-α production in human primary macrophages, with an IC50 of 9 μM. GSK-J4 hydrochloride serves as a cell-permeable proagent of GSK-J1 and is valuable for research into epigenetic regulation and inflammation-related pathways. -
KDM5 Inhibitor
KDM5-C70 is a potent and cell-permeable pan-KDM5 histone demethylase inhibitor, derived from KDM5-C49 as an ethyl ester. It exhibits significant antiproliferative activity in myeloma cells, effectively increasing global levels of H3K4me3. KDM5-C70 is valuable for research exploring epigenetic regulation and its implications in cancer biology. -
LSD1/KDM1A Inhibitor
GSK2879552 dihydrochloride is a selective and irreversible inhibitor of lysine specific demethylase 1 (LSD1/KDM1A). This compound exhibits potential antineoplastic activity, making it valuable in cancer research. Its ability to modify epigenetic regulation through LSD1 inhibition allows for exploration of therapeutic strategies in malignancies where LSD1 is implicated. -
KDM5 Inhibitor
KDOAM-25 citrate is a selective inhibitor of histone lysine demethylases 5 (KDM5) with IC50 values of 71 nM, 19 nM, 69 nM, and 69 nM for KDM5A, KDM5B, KDM5C, and KDM5D, respectively. This compound is known to enhance global H3K4 methylation at transcriptional start sites, thereby influencing gene expression. Its activity contributes to the inhibition of proliferation in multiple myeloma MM1S cells, making it a valuable tool for research in epigenetics and cancer biology. -
KDM5B Inhibitor
TK-129 is a potent inhibitor of KDM5B, exhibiting low toxicity and high affinity with an IC50 of 44 nM. By targeting KDM5B, TK-129 disrupts the KDM5B-associated Wnt signaling pathway, resulting in cardioprotective effects. This compound effectively reduces angiotensin II-induced activation of cardiac fibroblasts in vitro and mitigates isoprenaline-induced myocardial remodeling and fibrosis in vivo. TK-129 is valuable for research in cardiovascular disease mechanisms and therapeutic strategies. -
JMJD6 Inhibitor
JMJD6-IN-1 is a selective inhibitor of JMJD6, demonstrating an inhibition rate of 82% at a concentration of 10 μM. It effectively reduces the proliferation of MCF-7 and HCC4006 cancer cell lines, with IC50 values of 19.2 μM and 25.2 μM, respectively. This compound is suitable for research applications focused on cancer biology and the exploration of JMJD6’s role in tumorigenesis. -
KDM4C Inhibitor
KDM4C-IN-1 is a potent inhibitor of the lysine demethylase KDM4C, exhibiting an IC50 of 8 nM. This compound has demonstrated significant biological activity by inhibiting the proliferation of HepG2 and A549 cancer cell lines, with IC50 values of 0.8 µM and 1.1 µM, respectively. KDM4C-IN-1 is a valuable tool for research applications related to cancer biology and epigenetic regulation. -
KDM1A/LSD1 Inhibitor
DDP-38003 dihydrochloride is a novel orally bioavailable inhibitor of the lysine-specific demethylase 1A (KDM1A/LSD1) enzyme, demonstrating an IC50 of 84 nM. This compound is valuable for research applications involving epigenetic regulation and modulation of gene expression. Its inhibitory properties make it a pertinent tool for studies in cancer biology and potential therapeutic strategies targeting KDM1A/LSD1 pathways. -
LSD1 Enzyme Inhibitor
TAK-418 is a selective inhibitor of the lysine-specific demethylase 1 (LSD1, KDM1A) enzyme, exhibiting an IC50 of 2.9 nM. This compound modulates epigenetic regulation and has demonstrated efficacy in improving symptoms associated with autism in neurodevelopmental disorder models. Its ability to target LSD1 makes it a valuable tool for research into epigenetic mechanisms and potential therapeutic strategies for related disorders. -
KDM4A/KDM4B Inhibitor
NSC636819 is a competitive and selective inhibitor of the lysine demethylases KDM4A and KDM4B. By targeting these enzymes, NSC636819 may impede the progression of prostate cancer, thereby serving as a valuable tool in cancer research. Its application is particularly relevant for studies aimed at understanding the role of KDM4A/KDM4B in oncogenesis and potential therapeutic strategies for prostate cancer. -
KDM2B Inhibitor
KDM2B-IN-2 is a potent inhibitor of the histone demethylase KDM2B, demonstrating an IC50 of 0.021 μM in a KDM2B TR-FRET assay. This compound is utilized in research focused on hyperproliferative diseases, providing insights into epigenetic regulation and potential therapeutic applications. Its high specificity and efficacy make it a valuable tool for studying the role of KDM2B in various biological contexts. -
KDM5 Demethylases Inhibitor
KDM5-C49 hydrochloride is a potent and selective inhibitor of KDM5 demethylases, demonstrating IC50 values of 40 nM, 160 nM, and 100 nM for KDM5A, KDM5B, and KDM5C, respectively. This compound is instrumental for research investigating the role of KDM5 demethylases in cancer biology. Its specificity and inhibitory potency make KDM5-C49 hydrochloride a valuable tool for elucidating the mechanisms of cancer progression and potential therapeutic targets. -
LSD1 Inhibitor
Seclidemstat mesylate is a potent noncompetitive and reversible inhibitor of the lysine-specific demethylase 1A (LSD1/KDM1A), with a Ki of 31 nM and an IC50 of 13 nM. This compound enhances antitumor immunity in ovarian cancer associated with SWI/SNF complex mutations and demonstrates efficacy in inhibiting viral production, DNA replication, and late gene expression. Seclidemstat mesylate is suitable for research applications in Ewing Sarcoma and other malignancies involving LSD1 modulation. -
KDM5 Inhibitor
CPI-455 hydrochloride is a potent pan-KDM5 inhibitor, exhibiting an IC50 of 10 nM for KDM5A. This compound effectively inhibits KDM5 activity, leading to an increase in global levels of H3K4me3. It has demonstrated the ability to reduce the population of drug-tolerant persister cancer cells across various cancer cell line models subjected to standard chemotherapy or targeted treatments, making it a valuable tool for cancer research and therapeutic development. -
LSD Derivative
ALD-52 (1-Acetyl-LSD) serves as a prodrug for LSD, primarily targeting serotonin receptors 5-HT1A, 5-HT2A, and 5-HT2C, with binding affinities of 1054, 174, and 10.2 nM, respectively. Upon conversion to LSD, ALD-52 elicits a head-twitch response (HTR) in vivo, demonstrating its psychoactive properties. This compound is valuable in hallucinogen research, providing insights into its pharmacological effects and the underlying mechanisms of serotonergic activity. -
PARP10/PARP15 Inhibitor
PARP10/15-IN-3 is a dual inhibitor targeting PARP10 and PARP15, exhibiting IC50 values of 0.14 µM and 0.40 µM, respectively. This compound effectively penetrates cellular membranes and has demonstrated the ability to rescue cells from apoptosis. PARP10/15-IN-3 serves as a valuable tool for investigating the roles of PARP10 and PARP15 in cellular processes and offers potential applications in studies related to cancer therapy and cell survival mechanisms. -
JAK2/FLT3 Inhibitor
Flonoltinib TFA is a potent and orally bioavailable inhibitor that targets both JAK2 and FLT3, exhibiting IC50 values of 0.7 nM and 4 nM, respectively, alongside 26 nM and 39 nM for JAK1 and JAK3. This compound possesses significant anti-cancer activity, making it a valuable tool for research in oncology and therapeutic development against malignancies driven by these pathways. Its dual inhibition profile highlights its potential in addressing cancers associated with aberrant JAK2 and FLT3 signaling. -
Pim-1 Kinase Inhibitor
Pim-1 kinase inhibitor 2 specifically targets and inhibits Pim-1 kinase activity, a critical regulator of cell survival and proliferation. This compound has been demonstrated to induce apoptosis in various cell types, making it a valuable tool for cancer research. Its potent inhibitory effects on Pim-1 kinase provide insights into the molecular mechanisms of tumorigenesis and underscore its potential in therapeutic applications for cancer treatment. -
SIRT6 PROTAC Degrader
SZU-B6 is a SIRT6-protein-targeting chimeric degrader that achieves a DC50 of 45 nM and 154 nM in SK-HEP-1 and Huh-7 cell lines, respectively. It effectively inhibits the proliferation of SK-HEP-1 cells with an IC50 of 1.51 μM and suppresses colony formation in both SK-HEP-1 and Huh-7 cells. Additionally, SZU-B6 induces apoptosis and causes a cell cycle arrest in the G2/M phase in SK-HEP-1 cells, demonstrating notable antitumor efficacy in mouse models. This compound serves as a valuable tool for studying the functional roles of SIRT6 in cancer research. -
GSPT1/BRD4 Degrader
DP-15 is a targeted degrader for GSPT1 and BRD4, demonstrating DC50 values of 5.25 nM and 0.48 nM, respectively. This compound exhibits potent anti-proliferative effects against acute myeloid leukemia (AML) and non-Hodgkin lymphoma (NHL) cells, with IC50 values in the nanomolar range. Additionally, DP-15 induces G1 phase cell cycle arrest and promotes apoptosis in MOLM13 cells. In vivo studies have shown its effective anti-leukemia activity in MOLM-13 xenograft mouse models, supporting its potential application in cancer research. -
JAK2 Inhibitor
ZT55 is a potent and selective inhibitor of JAK2, exhibiting an IC50 value of 0.031 μM. This compound effectively inhibits the proliferation of JAK2V617F-expressing HEL cell lines, inducing apoptosis and cell cycle arrest. In vivo, ZT55 demonstrates significant efficacy in inhibiting the growth of HEL xenograft tumors in mouse models. It serves as a valuable tool for research in myeloproliferative neoplasms, including polycythemia vera and primary thrombocythemia. -
CDK6/PIM1 Inhibitor
CDK6/PIM1-IN-1 hydrochloride is a potent dual inhibitor targeting CDK6 and PIM1, exhibiting IC50 values of 39 nM and 88 nM, respectively, along with significant inhibition of CDK4 (IC50=3.6 nM). This compound effectively inhibits the proliferation of acute myeloid leukemia (AML) cells, induces G1 phase cell cycle arrest, and promotes apoptosis. CDK6/PIM1-IN-1 hydrochloride is a valuable tool for research investigating the role of CDK6 and PIM1 in cancer biology, particularly in the context of AML. -
HDAC Class I Inhibitor
HDAC-IN-27 dihydrochloride is a potent inhibitor of class I histone deacetylases (HDAC1-3) with IC50 values ranging from 0.43 to 3.01 nM. This compound displays significant antitumor activity both in vitro and in vivo, particularly against acute myeloid leukemia (AML) cell lines, through mechanisms that include apoptosis induction and increased histone acetylation (AcHH3 and AcHH4). HDAC-IN-27 dihydrochloride is an important tool for investigating the roles of HDACs in cancer biology, specifically within the context of AML research. -
SIRT1 Inhibitor
JGB1741 is a potent and selective inhibitor of SIRT1, exhibiting an IC50 of approximately 15 μM. It displays weak inhibitory effects on SIRT2 and SIRT3, with IC50 values greater than 100 μM. JGB1741 enhances the levels of acetylated p53, promoting p53-mediated apoptosis through modulation of the Bax/Bcl2 ratio, cytochrome c release, and PARP cleavage. This compound is valuable for research applications focusing on breast cancer. -
JAK2 Inhibitor
ON044580 is a potent non-ATP-competitive inhibitor of the JAK2 kinase, demonstrating IC50 values of 1.23 μM and 1.09 μM for wild-type and V617F mutant JAK2, respectively. This compound functions by either binding to the STAT-5 binding domain or an allosteric site of JAK2, leading to the suppression of JAK2 kinase activity. ON044580 effectively induces apoptosis in chronic myelogenous leukemia cells that exhibit resistance to Imatinib, and it also inhibits both wild-type and T315I mutant forms of the BCR-ABL kinase. This reagent holds promise for therapeutic applications in myeloproliferative disorders characterized by dysregulated JAK/STAT signaling. -
LSD1 Inhibitor
S2116 is a potent inhibitor of lysine-specific demethylase 1 (LSD1), derived from N-alkylated tranylcypromine (TCP). This compound enhances H3K9 methylation while concurrently promoting H3K27 deacetylation at super-enhancer regions. S2116 effectively induces apoptosis in TCP-resistant T-cell acute lymphoblastic leukemia (T-ALL) cells by downregulating the transcription of NOTCH3 and TAL1 genes, and it has demonstrated significant growth inhibition of T-ALL cells in xenotransplanted mouse models. This reagent holds potential for research applications in cancer biology and epigenetic regulation. -
JAK Inhibitor
Dehydrocrenatidine is a natural alkaloid that functions as a selective inhibitor of Janus kinases (JAK). This compound exhibits significant biological activity by inhibiting voltage-gated sodium channels, which may alleviate mechanical allodynia in neuropathic pain models. Dehydrocrenatidine serves as a valuable tool for research in pain mechanisms and the therapeutic targeting of JAK pathways. -
PARP10/PARP15 Inhibitor
PARP10/15-IN-2 is a potent dual inhibitor of PARP10 and PARP15, exhibiting IC50 values of 0.15 µM and 0.37 µM, respectively. This compound has demonstrated the ability to penetrate cellular membranes and effectively rescue cells from apoptosis. PARP10/15-IN-2 serves as a valuable tool for research into cell survival mechanisms and the modulation of PARP-related signaling pathways. -
HDAC/JAK/BRD4 Inhibitor
HDAC/JAK/BRD4-IN-1 is a potent inhibitor targeting histone deacetylases (HDAC), Janus kinases (JAK), and bromodomain-containing protein 4 (BRD4). This compound demonstrates significant anti-proliferative effects and promotes apoptosis in MDA-MB-231 breast cancer cells. Additionally, HDAC/JAK/BRD4-IN-1 exhibits promising anticancer activity in vivo, making it a valuable tool for research in cancer therapeutics and the study of epigenetic and signaling pathways. -
SMARCA2 Degrader
A947 is a selective SMARCA2 proteolysis-targeting chimera (PROTAC) that functions as a potent degrader of SMARCA2. It exhibits a binding affinity to the SMARCA2 bromodomain with a Kd value of 93 nM, establishing its effectiveness in mediating targeted protein degradation. This compound has significant applications in cancer research, facilitating studies on the role of SMARCA2 in tumorigenesis and potential therapeutic interventions. -
PARP/PI3K Inhibitor
PARP/PI3K-IN-1 is a potent inhibitor of both PARP and PI3K, exhibiting pIC50 values of 8.22 for PARP-1, 8.44 for PARP-2, and varying activity against PI3K isoforms with values of 8.25 for PI3Kα, 6.54 for PI3Kβ, 8.13 for PI3Kδ, and 6.08 for PI3Kγ. This compound demonstrates significant anticancer activity and is suitable for research applications targeting a variety of oncological disorders. Its dual inhibition may provide insights into therapeutic strategies for cancer treatment. -
LSD1 Inhibitor
S2157 is a potent inhibitor of lysine-specific demethylase 1 (LSD1), derived from N-alkylated tranylcypromine (TCP). It enhances H3K9 methylation while concurrently promoting H3K27 deacetylation at super-enhancer regions, contributing to its apoptotic effects in TCP-resistant T-cell acute lymphoblastic leukemia (T-ALL) cells through the repression of NOTCH3 and TAL1 gene transcription. Additionally, S2157 demonstrates efficient penetration of the blood-brain barrier, effectively eliminating CNS leukemia in mouse models transplanted with T-ALL cells, making it a valuable tool for cancer research. -
Sirtuin Inhibitor
Sirt1/2-IN-2 is a dual inhibitor targeting SIRT1 and SIRT2, exhibiting IC50 values of 1.8 μM and 2.4 μM, respectively. This compound effectively prevents the deacetylation of p53 while promoting acetylation of p53 and α-tubulin. Sirt1/2-IN-2 demonstrates pro-apoptotic properties and exhibits anti-proliferative effects on human leukemia cell lines, making it a valuable tool in cancer research and therapeutic studies targeting the sirtuin family. -
Aurora kinase Inhibitor
Aurora kinase-IN-8 is an orally active inhibitor of Aurora kinases, specifically targeting Aurora A and B kinases with IC50 values of 2.8 nM and 28.1 nM, respectively. This compound effectively disrupts spindle formation, induces G2/M phase arrest, and promotes apoptosis in cancer cells. It is particularly relevant for research applications focused on malignancies, including triple-negative breast cancer. -
Pim-1 Inhibitor
Pim-1 kinase inhibitor 8 is a selective inhibitor of the PIM-1 kinase, exhibiting an IC50 value of 14.3 nM. This compound effectively disrupts cellular proliferation and migration by inhibiting PIM-1, leading to the induction of apoptosis and autophagy. In vivo studies demonstrate its capability to inhibit solid tumor growth in Solid Ehrlich Carcinoma (SEC)-bearing mice. Pim-1 kinase inhibitor 8 is valuable for research focused on breast and liver cancer. -
LSD1 Inhibitor
Geranylgeranoic acid is a Lysine-specific demethylase 1 (LSD1) inhibitor, exhibiting an IC50 value of 46.97 µM. This isoprenoid compound has demonstrated significant apoptosis-inducing properties through the disruption of mitochondrial membrane potential and the activation of caspase pathways, specifically interleukin-1β-converting enzyme (ICE) and cysteine protease precursor 32 (CPP32). Geranylgeranoic acid is applicable in cancer research and is derived from S. chinensis, highlighting its potential as an anticancer agent in studies involving human hepatoma cells and mouse hepatocytes. -
HDAC1-3 Inhibitor
HDAC-IN-53 is a selective inhibitor of histone deacetylases 1-3, demonstrating IC50 values of 47 nM, 125 nM, and 450 nM for HDAC1, HDAC2, and HDAC3, respectively. This compound exhibits minimal off-target effects, as it does not inhibit class II HDACs (IC50 > 10 μM). HDAC-IN-53 promotes caspase-dependent apoptosis and has been shown to inhibit the growth of human tumor xenografts in nude mice, as well as murine tumors in immune-competent mice bearing MC38 colon cancer. It serves as a valuable tool for studying cancer biology and potential therapeutic strategies targeting HDAC pathways. -
CDK6/PIM1 Inhibitor
CDK6/PIM1-IN-1 is a potent dual inhibitor targeting CDK6 and PIM1, with IC50 values of 39 nM and 88 nM, respectively, and an additional inhibition of CDK4 at an IC50 of 3.6 nM. This reagent significantly inhibits the proliferation of acute myeloid leukemia (AML) cells, induces G1 phase cell cycle arrest, and promotes apoptosis. CDK6/PIM1-IN-1 demonstrates strong anti-AML activity, making it a valuable tool for research in cancer biology and therapeutic development. -
HDAC6 Inhibitor
QTX125 TFA is a potent and highly selective inhibitor of Histone Deacetylase 6 (HDAC6). This compound demonstrates exceptional selectivity for HDAC6 over other isoforms, making it a valuable tool for studying the role of HDAC6 in various biological processes. QTX125 TFA has shown promising antitumor effects, indicating its potential for use in cancer research and therapeutic applications targeting HDAC6-related pathways. -
CBP Inhibitor
DC-CPin711 is a potent and selective inhibitor of the CREB-binding protein (CBP) bromodomain, demonstrating an IC50 of 0.0626 μM. This compound effectively induces apoptosis and arrests the cell cycle at the G1 phase, making it a valuable tool for research into cellular proliferation and death pathways. Its specificity for CBP enhances its utility in investigating the role of bromodomain-containing proteins in various biological processes and diseases. -
HDAC Inhibitor
CRA-026440 hydrochloride is a potent, broad-spectrum histone deacetylase (HDAC) inhibitor, exhibiting Ki values against recombinant HDAC isoenzymes of 4 nM for HDAC1, 14 nM for HDAC2, 11 nM for HDAC3, 15 nM for HDAC6, 7 nM for HDAC8, and 20 nM for HDAC10. This compound demonstrates significant antitumor and antiangiogenic activities, making it relevant for studies in cancer biology. Additionally, CRA-026440 hydrochloride possesses an alkyne functional group, enabling it to participate in copper-catalyzed azide-alkyne cycloaddition (CuAAc), facilitating its use in click chemistry applications for bioconjugation studies. -
PARP1 Inhibitor
KU-0058948 is a potent inhibitor of PARP1, exhibiting an IC50 value of 3.4 nM. This compound induces cell cycle arrest and apoptosis in primary myeloid leukemic cells as well as established myeloid leukemic cell lines. It is suitable for research applications focused on cancer biology and the exploration of PARP1's role in cellular processes.
