Epigenetics
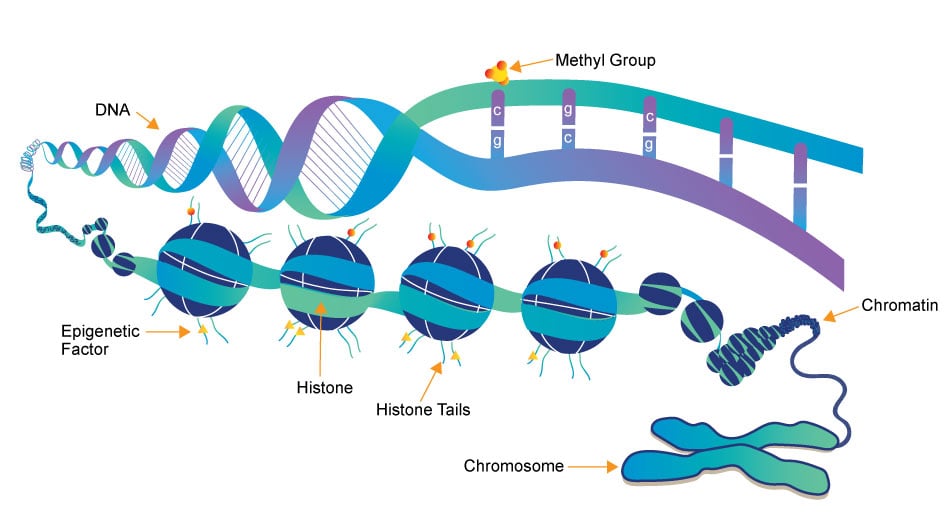
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
PROTAC BRD4 Degrader
PROTAC BRD4 Degrader-8 is a proteolysis-targeting chimera (PROTAC) that engages both von Hippel-Lindau (VHL) and BRD4, exhibiting IC50 values of 1.1 nM for BRD4 BD1 and 1.4 nM for BRD4 BD2. This compound effectively promotes the degradation of BRD4 protein in PC3 prostate cancer cells, making it a valuable tool for research into BRD4-related signaling pathways and cancer therapeutics. Its high potency highlights its potential use in studying mechanisms of protein regulation and developing novel cancer treatments. -
PROTAC BRD Degrader
β-NF-JQ1 is a PROTAC that targets bromodomain-containing (BRD) proteins by leveraging β-NF as a ligand for the Aryl Hydrocarbon Receptor (AhR) E3 ligase. This compound facilitates the degradation of BRD proteins through the recruitment of AhR, leading to effective protein knockdown. β-NF-JQ1 demonstrates significant anticancer activity, making it a valuable tool for research in cancer biology and the development of targeted therapeutic strategies. -
CBP/p300 PROTAC Degrader
CBPD-268 is a highly potent CBP/p300 PROTAC degrader, exhibiting a DC50 value of ≤ 0.03 nM. This compound effectively induces degradation of CBP/p300 and demonstrates significant inhibition of cell growth, showcasing its antitumor potential. CBPD-268 is particularly relevant for research into androgen receptor-positive prostate cancer, facilitating studies on novel therapeutic strategies. -
Isoform of SHP2-D26
SHP2-D26 isomer-1 is an isoform of the SHP2-D26 degrader, designed specifically for targeted protein degradation applications. This compound does not induce degradation of SHP2 at concentrations ranging from 3 to 1000 nM. As a component of PROTAC technology, SHP2-D26 isomer-1 serves as a valuable tool for studying SHP2's role in signaling pathways and assessing the therapeutic potential of SHP2 modulation in various diseases. -
EZH2 PROTAC Degrader
MS8847 is a powerful EZH2 PROTAC degrader that utilizes the E3 ligase von Hippel-Lindau (VHL) to promote the degradation of EZH2 through the ubiquitin-proteasome system. This compound demonstrates robust anti-proliferative effects in acute myeloid leukemia (AML) and triple-negative breast cancer (TNBC) cell lines. MS8847 serves as a valuable tool for investigating the role of EZH2 in oncogenic signaling pathways and developing targeted therapies for EZH2-dependent malignancies. -
PROTAC PARP1 Degrader
PROTAC PARP1 Degrader functions as a targeted protein degradation agent that specifically targets PARP1 through the MDM2 E3 ligase. This compound effectively induces PARP1 cleavage and promotes apoptosis in cancer cells, making it valuable for research in cancer therapies. The structure includes the MDM2 ligand, the PARP1 ligand, and a PEG-based linker, facilitating efficient delivery and degradation of the target protein. -
PROTACs
PROTAC BRD9 Degrader-4 is a bifunctional degrader targeting BRD9 through the proteolysis-targeting chimera (PROTAC) mechanism. It effectively induces degradation of BRD9, thereby altering oncogenic pathways associated with various cancers. This compound is utilized in cancer research to study the therapeutic potential of BRD9 modulation and the implications of targeted degradation in tumor biology. -
BRD42/BRD4 Degrader
IBG1 is a molecular glue degrader that selectively targets BRD2 and BRD4, exhibiting a degradation capability with a DC50 of 0.15 nM. Notably, IBG1 does not significantly impact the paralogue BRD3. This compound effectively inhibits the growth of cancer cells and is a valuable tool for research focused on tumor biology and therapeutic strategies. -
SMARCA2 PROTAC degrader
SMD-3040 is a selective SMARCA2 PROTAC degrader with a DC50 of 12 nM and a maximal degradation efficiency of 91%. This compound effectively inhibits tumor cell proliferation, demonstrating significant antitumor activity. SMD-3040 is particularly useful in studies related to various tumors, including melanoma. -
PROTAC BRD4 Degrader
PROTAC BET Degrader-10 is a selective degrader targeting the BET protein BRD4 through a PROTAC mechanism, featuring a DC50 of 49 nM. This compound utilizes ligands for Cereblon and BRD4, promoting the ubiquitination and subsequent degradation of BRD4. It serves as a valuable tool for investigating the role of BRD4 in various biological processes and cancer-related research applications. -
JAK1 PROTAC Degrader
PROTAC JAK1 Degrader 1 is a selective PROTAC agent targeting JAK1, exhibiting a DC50 of 214 nM. This compound initiates rapid degradation of JAK1, leading to significant antitumor activity. It serves as a valuable tool for investigating JAK1-related signaling pathways and developing therapeutic strategies for malignancies driven by JAK1 dysregulation. -
PROTAC BRD4 Degrader
PROTAC BRD4 Degrader-1 is a novel PROTAC that utilizes ligands for both Cereblon and BRD4, exhibiting an IC50 of 41.8 nM against the BRD4 bromodomain 1 (BD1). This compound promotes the degradation of BRD4 protein, leading to a significant decrease in c-Myc expression. It is designed for research applications aimed at understanding the role of BRD4 in transcriptional regulation and its implications in various cancers. -
PROTAC EED Degrader
PROTAC EED Degrader-2 is a von Hippel-Lindau-based PROTAC that selectively targets the embryonic ectoderm development (EED) subunit of the polycomb repressive complex 2 (PRC2), exhibiting a pKD of 9.27. This compound acts as an effective inhibitor, demonstrating a pIC50 of 8.11, facilitating the degradation of EED and providing valuable insights for research on PRC2-related epigenetic regulation and its implications in various biological processes. Applications include studies on gene expression regulation and potential therapeutic targets in cancer and developmental biology. -
PROTAC BRD4 Degrader
KB02-JQ1 is a selective PROTAC BRD4 degrader that functions as a molecular glue, specifically targeting BRD4 while sparing BRD2 and BRD3. This compound induces BRD4 degradation by covalently modifying the E3 ligase DCAF16, thereby enhancing the stability and duration of protein degradation in biological systems. Its unique design, which incorporates JQ1 linked to the ubiquitin E3 ligase ligand KB02, facilitates targeted modulation of gene expression, making it a valuable tool for research in cancer biology and therapeutic development. -
TYK2 Degrader
PROTAC TYK2 degradation agent1 is a selective degrader targeting TYK2, effectively facilitating its degradation with a DC50 value of 14 nM. This compound highlights significant biological activity in modulating TYK2 levels, making it a useful tool for investigating autoimmune diseases. Research applications include studying the role of TYK2 in inflammatory pathways and evaluating potential therapeutic strategies. -
PROTAC EED Degrader
PROTAC EED degrader-1 is a von Hippel-Lindau-based PROTAC that selectively targets EED with a pKD of 9.02. This compound functions as a polycomb repressive complex 2 (PRC2) inhibitor, exhibiting an inhibitory potency characterized by a pIC50 of 8.17. It serves as a valuable tool for researching the modulation of PRC2 activity and its implications in cancer biology and epigenetic regulation. -
SIRT2 Inhibitor
SIRT2-IN-8 is a selective inhibitor of SIRT2 (Sirtuin 2), a member of the sirtuin family of proteins implicated in various cellular processes. This compound exhibits strong inhibition of SIRT2 activity, making it a valuable tool for investigating the role of SIRT2 in neurodegenerative diseases, particularly Huntington's and Parkinson's diseases. Its use in research can contribute to a better understanding of the molecular mechanisms underlying these conditions and aid in the development of therapeutic strategies. -
HDAC Inhibitor
HC-Toxin is a potent histone deacetylase (HDAC) inhibitor with an IC50 of 30 nM. This cyclic tetrapeptide effectively induces apoptosis in tumor cells, demonstrating significant anticancer activity. Its mechanism of action makes it valuable for research in cancer therapy and the modulation of gene expression. -
SIRT Inhibitor
Nicotinamide is a form of vitamin B3 or niacin. Nicotinamide Hydrochloride inhibits SIRT2 activity (IC50: 2 μM). Nicotinamide also inhibits SIRT1. Nicotinamide increases cellular NAD+, ATP, ROS levels. Nicotinamide inhibits tumor growth and improves survival. Nicotinamide also has anti-HBV activity. -
PTPN1/PTPN2 Inhibitor
Osunprotafib (ABBV-CLS-484) is an orally active and selective active site PTPN1 (IC50: 2.5 nM) and PTPN2(IC50: 1.8 nM) inhibitor. Osunprotafib has 6-8-fold weaker activity on PTPN9 and no detectable activity on SHP-1 or SHP-2. Osunprotafib increases the sensitivity of human cancer cell lines to IFNγ. Osunprotafib generates robust anti-tumor immunity by enhancing JAK-STAT signalling and reducing T cell dysfunction. -
Antiepileptic Agent
Levetiracetam is an antiepileptic agent that targets the synaptic vesicle protein SV2A. It has been shown to enhance the effects of Temozolomide on glioblastoma stem cell proliferation and apoptosis. Additionally, Levetiracetam modulates histone deacetylase (HDAC) levels, leading to the silencing of MGMT, which further improves the efficacy of Temozolomide. This compound serves as a valuable chemosensitizer in cancer research applications. -
PDE Inhibitor
Theophylline, a potent phosphodiesterase (PDE) inhibitor, primarily targets PDE3, leading to relaxation of airway smooth muscle and enhanced bronchodilation. This compound also functions as an adenosine receptor antagonist and exhibits anti-inflammatory properties by elevating IL-10 levels and inhibiting NF-κB translocation into the nucleus. Additionally, Theophylline has been shown to induce apoptosis in certain cell types. Its applications are particularly relevant in the research of asthma and chronic obstructive pulmonary disease (COPD). -
DNA Methyltransferase Inhibitor
γ-Oryzanol is an effective inhibitor of DNA methyltransferases (DNMTs) with a primary focus on DNMT1 and DNMT3a. It demonstrates significant inhibitory activity, with an IC50 of 3.2 μM for DNMT1 and 22.3 μM for DNMT3a. This compound has important implications for epigenetic research and may be useful in studies exploring gene expression regulation and potential therapeutic applications in cancer and other diseases associated with aberrant DNA methylation. -
PIM2 Inhibitor
JP-11646 is a potent pan-PIM inhibitor specifically targeting PIM2 with an IC50 of 0.5 nM. This reversible, ATP non-competitive inhibitor significantly reduces the mRNA levels of PIM1, PIM2, and PIM3. JP-11646 has demonstrated efficacy in inhibiting cell viability in small cell lung cancer (SCLC) and large cell neuroendocrine carcinomas of the lung (LCNEC), leading to apoptosis or necroptosis through decreased p-4EBP-1 and altered caspase activity. This reagent is valuable for research applications in SCLC, LCNEC, acute myeloid leukemia (AML), multiple myeloma (MM), and triple-negative breast cancer (TNBC). -
BRD4 Degrader
PROTAC BRD4 Degrader-6 is a potent small-molecule degrader that targets BRD4, exhibiting an IC50 value of 2.7 nM for the BRD4 BD1 domain. This compound effectively degrades BRD4 protein and leads to the downregulation of c-Myc expression. In vitro studies demonstrate its capacity to inhibit proliferation and induce apoptosis in the pancreatic cancer cell line BxPC3, making it a valuable tool for research in human pancreatic cancer biology. -
PIM-1/2 Inhibitor
Pim-1 kinase inhibitor 10 is a selective inhibitor of PIM-1 and PIM-2 kinases, functioning through both competitive and non-competitive mechanisms. This compound effectively induces apoptosis in cancer cells, demonstrating significant anticancer activity. Additionally, Pim-1 kinase inhibitor 10 activates caspase 3 and 7, further contributing to its potential as a therapeutic agent in cancer research. -
HDAC3 Inhibitor
HDAC3-IN-2 is a potent inhibitor of histone deacetylase 3 (HDAC3), with an IC50 value of 14 nM. This pyrazinyl hydrazide compound exhibits cytotoxicity against triple-negative breast cancer cell lines, demonstrating an IC50 of 0.55 μM for 4T1 cells and 0.74 μM for MDA-MB-231 cells. In in vivo studies using tumor-bearing mouse models, HDAC3-IN-2 effectively enhances histone acetylation levels at H3K9, H3K27, and H4K12 while promoting apoptosis through increased caspase-3, caspase-7, and cytochrome c levels, alongside a decrease in proliferation markers such as Bcl-2, CD44, EGFR, and Ki-67. -
PARP1 Inhibitor
PARP-1-IN-2 is a potent inhibitor of PARP1, exhibiting an IC50 value of 149 nM. This compound demonstrates significant anti-proliferative effects on the A549 human lung adenocarcinoma epithelial cell line and induces apoptosis in these cells. Its favorable ADME profile suggests high permeability across the blood-brain barrier, making it a valuable tool for research in cancer biology and therapeutic applications targeting PARP1-related pathways. -
JAK-STAT Inhibitor
WP-1034 is a selective JAK-STAT inhibitor that exhibits pro-apoptotic and antileukemic properties, particularly in acute myeloid leukemia (AML) models. By blocking the activation of Stat 3 and Stat 5, WP-1034 effectively induces cell cycle arrest and triggers apoptosis in affected cells. This reagent is valuable for research focused on understanding the mechanisms and therapeutic avenues in AML. -
KDM1/CDK1 Inhibitor
KDM1/CDK1-IN-1 is a potent inhibitor of both KDM1 and CDK1, exhibiting IC50 values of 0.096 μM and 0.078 μM, respectively. This compound effectively induces cell cycle arrest at the G2/M phase and promotes apoptosis in HOP-92 cancer cells. Additionally, KDM1/CDK1-IN-1 demonstrates significant cytotoxic effects against a range of cell lines, including CCRF-CEM, HOP-92, and Hep-G2, with IC50 values of 16.34 μM, 3.45 μM, and 7.79 μM, respectively. Its ability to target critical regulators of the cell cycle makes KDM1/CDK1-IN-1 valuable for cancer research applications. -
PARP1 Inhibitor
4,4′-Secalonic acid D is a potent inhibitor of PARP1, a key enzyme in the DNA repair pathway. This compound promotes the accumulation of reactive oxygen species (ROS) and DNA damage, leading to the activation of the caspase-3/GSDME pathway, which triggers apoptosis and pyroptosis in tumor cells. 4,4′-Secalonic acid D exhibits significant anti-tumor activity, making it a valuable tool for cancer research and therapeutic investigations. -
HDAC1/6 Inhibitor
HDAC1/6-IN-3 is a potent inhibitor of histone deacetylases 1 and 6 (HDAC1 and HDAC6). It demonstrates strong inhibitory activity, with IC50 values of 1.1 nM for HDAC1 and 2.7 nM for HDAC6. This compound effectively induces cell cycle arrest in the G0/G1 phase and promotes both apoptosis and pyroptosis in HepG2 cells. Additionally, HDAC1/6-IN-3 exhibits significant antitumor effects in the HepG2 xenograft model and is valuable for research focused on various types of cancer, including liver, lung, colon, and breast cancers. -
PARP-1 Inhibitor
PARP-1-IN-3 is a potent inhibitor of PARP-1, with IC50 values of 0.25 nM for PARP-1 and 2.34 nM for PARP-2. This benzamide derivative effectively induces apoptosis and leads to G2/M phase cell cycle arrest. PARP-1-IN-3 is valuable for research applications focused on cancer mechanisms and therapeutic strategies. -
LSD1/ DCN1-UBC12 Protein-Protein Interaction Inhibitor
WS-384 is a dual inhibitor targeting LSD1 and the DCN1-UBC12 protein-protein interaction, demonstrating IC50 values of 338.79 nM and 14.81 nM, respectively. This compound exhibits significant anticancer activity, facilitating cell cycle arrest, DNA damage, and apoptosis in cancer cells. WS-384 serves as a valuable tool for research into non-small cell lung cancer (NSCLC) and other related malignancies. -
histone acetyltransferase Gcn5 inhibitor
Butyrolactone 3 (MB-3) is a specific small-molecule inhibitor of the histone acetyltransferase Gcn5, exhibiting an IC₅₀ of 100 μM and binding affinity comparable to that of its natural substrate, histone H3. It displays weak inhibitory activity against CBP (IC₅₀ = 0.5 mM), indicating high selectivity for Gcn5. By modulating histone acetylation and epigenetic regulation, Butyrolactone 3 serves as a valuable research tool for exploring the roles of Gcn5 in cancer, metabolic disorders, autoimmune diseases, and neurological conditions. -
KDM4D Inhibitor
Zavondemstat (QC8222; TACH 101) is an inhibitor of histone lysine demethylase 4D (KDM4D) with demonstrated antineoplastic activity. It is under investigation for its potential use in cancer therapy by targeting epigenetic regulation mechanisms. - Tz-Thalidomide is a tetrazine-tagged thalidomide derivative that functions as a ligand for E3 ligases. It exhibits binding affinity for BRD4, with IC₅₀ values of 46.25 μM for BRD4-1 and 62.55 μM for BRD4-2. As a click chemistry reagent, Tz-Thalidomide contains a tetrazine moiety capable of undergoing inverse electron demand Diels–Alder (iEDDA) reactions with trans-cyclooctene (TCO)-containing molecules, enabling bioorthogonal labeling and conjugation applications.
-
HDAC inhibitor
Nullscript is an inactive analog of Scriptaid and serves as a negative control for Scriptaid, a representative histone deacetylase (HDAC) inhibitor. Despite its inactivity as an HDAC inhibitor, Nullscript inhibits the growth of *Cryptosporidium parvum* with an IC₅₀ value of 2.1 μM. -
SMARCA4/SMARCA2 ATPase inhibitor
FHD-286 is a selective, orally active inhibitor of the SMARCA4/SMARCA2 (BRG1/BRM) ATPase. It holds potential for research into BAF (BRG1/BRM-associated factor)-related disorders, including acute myeloid leukemia. -
p300/CBP inhibitor
NEO2734 (EP31670) is an orally active dual inhibitor of p300/CBP and BET bromodomains, with IC₅₀ values of <30 nM for both targets. It is effective in both SPOP-mutant and wild-type prostate cancer models. -
SMARCA4/SMARCA2 ATPase Inhibitor
FHT-1015 is a selective allosteric inhibitor of SMARCA4 (BRG1) and SMARCA2 (BRM), with IC₅₀ values of 4 nM and 5 nM, respectively. It binds to an allosteric site, inducing conformational changes that inhibit the ATPase activity of BRG1/BRM. FHT-1015 disrupts tumor cell growth and migration and is applicable in research on uveal melanoma and hematologic malignancies. -
ATAD2 bromodomain inhibitor
GSK8814 is a potent and selective chemical probe and inhibitor of the ATAD2 bromodomain, with an IC₅₀ of 0.059 μM, a pK\_d of 8.1, and a pK\_i of 8.9 in BROMOscan. It binds to ATAD2 and BRD4 BD1 with pIC₅₀ values of 7.3 and 4.6, respectively, demonstrating over 500-fold selectivity for ATAD2. GSK8814 is suitable for research in cancers associated with ATAD2 bromodomain activity. - NICE-01 (AP1867-PEG2-JQ1; AP-PEG2-JQ1) is a bifunctional compound designed to induce nuclear import of cytosolic proteins. It functions by binding proteins in distinct cellular compartments and leveraging nuclear-localized BRD4 as a “carrier” to facilitate co-import and nuclear retention of cytosolic cargoes.
-
Menin inhibitor
BN-104 (BNM-1192) is an orally active and selective brain-penetrant menin inhibitor that disrupts the menin-MLL interaction, leading to degradation of the menin protein. It exhibits antitumor activity and is applicable in cancer research, including studies on acute myeloid leukemia. BN-104 is a weak hERG inhibitor, with an IC₅₀ greater than 100 μM. -
Menin inhibitor
Enzomenib is an inhibitor of menin, a protein encoded by the multiple endocrine neoplasia (MEN) gene. It disrupts the interaction between menin and mixed lineage leukemia (MLL) fusion proteins and is applicable in the study of hematological malignancies. -
BET/EP300 inhibitor
XP-524 is a potent dual inhibitor of BET and EP300, exhibiting strong antitumor activity in vivo. It prevents KRAS-induced neoplastic transformation and prolongs survival in two transgenic mouse models of aggressive pancreatic ductal adenocarcinoma (PDAC). XP-524 also enhances self-peptide presentation and promotes tumor infiltration by cytotoxic T lymphocytes, supporting its potential for PDAC research. -
CBP/p300 inhibitor
CBP/p300-IN-8 is a potent inhibitor of the CBP/p300 family of bromodomains, with an IC₅₀ of 0.01–0.1 µM for CBP. It also inhibits BRD4 activity with significantly lower potency (IC₅₀ = 1–1000 µM).
