Epigenetics
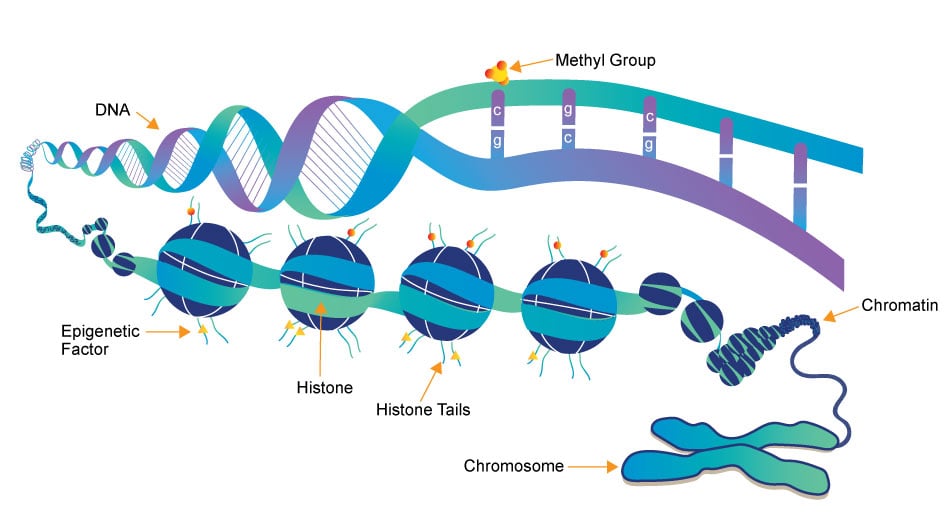
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
JAK1 Inhibitor
JAK1-IN-12 is a selective inhibitor of Janus kinase 1 (JAK1), exhibiting an IC50 of 0.0246 μM, and demonstrating weaker inhibition of JAK2, JAK3, and TYK2 with IC50 values of 0.423 μM, 0.410 μM, and 1.12 μM, respectively. This compound has been shown to promote hair growth in murine models. JAK1-IN-12 is a valuable tool for investigating immune and inflammatory diseases, providing insights into JAK1-mediated signaling pathways. -
Tyk2 Inhibitor
Tyk2-IN-23 is a selective TYK2 inhibitor with an IC50 of 18 nM, demonstrating over 70-fold selectivity against JAK isoforms 1, 2, and 3. This compound effectively inhibits phosphorylated STAT3 in TYK2-dependent signaling pathways activated by interferon-alpha (IFN-α) and interleukin-10 (IL-10). Additionally, Tyk2-IN-23 significantly reduces IFN-α-induced STAT1 phosphorylation in H9 cells, making it a valuable tool for the investigation of alopecia areata and allergic rhinitis. -
PTPN2/1 Dual Inhibitor
PTPN2/1-IN-4 is a potent dual inhibitor of PTPN1 and PTPN2, exhibiting IC50 values of 12.8 nM and 5.8 nM, respectively. This compound effectively modulates the IFNγ-JAK-STAT signaling pathway, resulting in enhanced CD8+ T-cell infiltration into tumors. PTPN2/1-IN-4 demonstrates significant anticancer activity, inhibiting tumor growth both as a standalone treatment and in combination with anti-PD-1 antibodies in B16-OVA syngeneic mouse models, making it a valuable tool for cancer research. -
JAK1 Inhibitor
JAK1-IN-9 is a potent and selective inhibitor of Janus kinase 1 (JAK1), with an IC50 of 72 nM. This compound demonstrates notable selectivity over other JAK family members, exhibiting an inhibition of 12-fold or more. JAK1-IN-9 is valuable for research applications focused on unraveling the roles of JAK1 in cellular signaling pathways and the development of therapeutic strategies for diseases involving JAK1 dysregulation. -
TYK2/JAK2 Inhibitor
Soficitinib is a selective inhibitor of tyrosine kinase 2 (TYK2) and Janus kinase 2 (JAK2), exhibiting IC50 values of 0.5 nM and 1.2 nM, respectively. This compound is primarily utilized in research related to autoimmune diseases, such as psoriasis and rheumatoid arthritis, as well as inflammatory conditions. Soficitinib's potent inhibition profiles make it a valuable tool for studying the underlying mechanisms of these diseases and evaluating potential therapeutic strategies. -
JAK1/2 Ligand
JAK1/2 Ligand 1 is a selective ligand targeting the Janus kinases JAK1 and JAK2. This compound is essential for the development of Proteolysis Targeting Chimeras (PROTACs) and contributes to the synthesis of JAK1/2 Ligand-Linker Conjugates. It provides a valuable tool for studying the JAK signaling pathway and examining its role in various biological processes, including immunity and inflammation. -
JAK3 Inhibitor
MS-1020 is a potent ATP-competitive inhibitor of JAK3, targeting the JAK3/STAT signaling pathway. By inhibiting JAK3, MS-1020 effectively induces apoptosis and promotes cell death, alongside decreasing levels of tyrosine phosphorylated STAT3. This compound holds potential for research applications focused on cancers associated with dysregulated JAK3 signaling. -
JAK3/BTK Inhibitor
JAK3/BTK-IN-5 is a potent dual inhibitor targeting Janus kinase 3 (JAK3) and Bruton's tyrosine kinase (BTK). This compound effectively disrupts the BTK/JAK3 signaling pathway, demonstrating potential synergistic effects advantageous for the treatment of autoimmune diseases. JAK3/BTK-IN-5 is valuable for research into conditions related to JAK3 kinase and BTK, making it a crucial tool for studying the pathophysiology of these targets. -
FLT3/JAK2/BRD4 Ligand
FLT3/JAK2/BRD4 ligand-1 is a specific ligand for the FLT3, JAK2, and BRD4 proteins. This compound facilitates the design and synthesis of proteolysis-targeting chimeras (PROTACs), specifically PROTAC FLT3/JAK2/BRD4 Degrader-1. It demonstrates potential applications in targeted protein degradation and cancer research, enabling innovative therapeutic strategies against malignancies associated with these targets. -
JAK2 Inhibitor
JAK2-IN-9 is a selective JAK2 inhibitor with an IC50 of 5 nM, effectively targeting and inhibiting the phosphorylation of JAK2, as well as downstream signaling proteins STAT3 and STAT5. This compound exhibits metabolic stability and induces apoptosis in targeted cells. JAK2-IN-9 is valuable for research focused on myeloproliferative neoplasms (MPNs) and contributes to elucidating the role of JAK2 signaling in hematological malignancies. -
JAK3/TEC Inhibitor
JAK3-IN-18 is a potent and selective inhibitor of JAK3 and TEC, exhibiting IC50 values of 0.5391 nM and 12.40 nM, respectively. With over 10,000-fold selectivity against AK1, AK2, and TYK2, JAK3-IN-18 shows exceptional efficacy in preclinical models of autoimmune disorders, particularly in the experimental autoimmune encephalomyelitis (EAE) mouse model. This compound is valuable for research applications focused on multiple sclerosis and related autoimmune conditions. -
JAK Inhibitor
JAK-IN-15 is a Janus kinase (JAK) inhibitor that selectively targets JAK family members involved in the signaling pathways of various cytokines. This compound demonstrates significant anti-inflammatory activity and is primarily utilized in research applications focused on autoimmune diseases and hematological malignancies. JAK-IN-15 serves as a valuable tool for studying JAK-mediated signal transduction and exploring potential therapeutic strategies targeting JAK-related disorders. -
Tyk-2 Inhibitor
Tyk2-IN-9 is a selective inhibitor of the tyrosine kinase 2 (Tyk2) with IC50 values of 0.076 nM for TYK2-JH2 and 1.8 nM for JAK1-JH2. This compound demonstrates potent inhibition of Tyk2 activity, making it a valuable tool for investigating the mechanisms underlying inflammatory and autoimmune diseases. Its specificity may enhance research efforts aimed at developing targeted therapies in these areas. -
Tyk2 Inhibitor
Tyk2-IN-22 is a selective inhibitor of tyrosine kinase 2 (Tyk2) with a notable inhibition profile, exhibiting IC50 values of 9.7 nM, 148.6 nM, and 883.3 nM against Tyk2, JAK1, and JAK3, respectively. This compound effectively inhibits downstream phosphorylation of STAT5, making it a valuable tool for studying Tyk2-mediated signaling pathways. Tyk2-IN-22 is suitable for research applications related to autoimmune diseases, inflammatory conditions, and cancer therapies targeting JAK-STAT signaling. -
JAK Inhibitor
(Rac)-TUL01101 is a selective inhibitor of Janus kinases (JAK). This compound plays a significant role in modulating cytokine signaling pathways, making it valuable in the study of inflammatory diseases. Its effectiveness in research applications includes investigations into conditions such as rheumatoid arthritis, atopic dermatitis, and alopecia areata. -
JAK/TYK2 Inhibitor
JAK1/TYK2-IN-4 is a dual inhibitor targeting JAK1 and TYK2, demonstrating IC50 values of 39 nM and 21 nM, respectively. This compound is notable for its oral bioavailability and has potential applications in the study of cytokine signaling and immune responses. It is particularly relevant for research in autoimmune diseases and hematological disorders, providing a valuable tool for elucidating the role of JAK/TYK2 pathways in various biological contexts. -
BFAR Inhibitor
iBFAR2 is a selective inhibitor of the BFAR protein, targeting its role in immune modulation. This compound effectively restores the CD8+ tissue-resident memory T (TRM) cell subset, enhancing anti-tumor immunity in solid tumors. iBFAR2 promotes the association between JAK2 and STAT1 as well as the subsequent phosphorylation of STAT1, contributing to its biological activity in cancer research applications. -
JAK2/STAT3 Iinhibitor
HD-2a is a JAK2/STAT3 inhibitor that functions by downregulating circDcbld2 expression in RAW264.7 cells. This compound is valuable for research into the modulation of the JAK2/STAT3 signaling pathway, which is implicated in various inflammatory and autoimmune disorders. HD-2a can aid in the exploration of therapeutic strategies targeting JAK2/STAT3-mediated processes. -
TYK2 Inhibitor
TYK2 activator-1 is a selective TYK2 activator with an EC50 value of 1.78 μM. This compound also inhibits JAK2 and JAK3, exhibiting IC50 values of 6.8 μM and 6.3 μM, respectively. Due to its unique profile, TYK2 activator-1 is suited for research applications targeting cytokine signaling pathways and immune responses, particularly in the context of autoimmune diseases and inflammatory disorders. -
BTK/JAK3 Inhibitor
JAK3/BTK-IN-6 is a potent dual inhibitor targeting Bruton's Tyrosine Kinase (BTK) and Janus Kinase 3 (JAK3), exhibiting IC50 values of 0.6 nM and 0.4 nM, respectively. This compound demonstrates excellent metabolic stability in human liver microsomes, making it suitable for in vitro studies. JAK3/BTK-IN-6 is valuable in research applications related to hematological disorders and immune system dysfunctions. -
JAK Inhibitor
JAK-IN-27 is a potent inhibitor of the Janus kinase (JAK) family, demonstrating IC50 values of 3.0 nM for TYK2, 7.7 nM for JAK1, and 629.6 nM for JAK3. This compound effectively inhibits IFN-α2B-induced phosphorylation of STAT3 in Jurkat cells, with an IC50 of 23.7 nM. JAK-IN-27 is valuable for research applications involving JAK signaling pathways and their role in immune responses and various inflammatory diseases. -
JAK Inhibitor
TK4g is a selective Janus kinase (JAK) inhibitor, exhibiting IC50 values of 12.61 nM for JAK2 and 15.80 nM for JAK3. This compound is crucial for researching lymphoid-derived diseases and various leukemias, providing insights into the role of JAK signaling in cancer biology. -
Tyk-2 Inhibitor
TYK2-IN-11 is a selective inhibitor of Tyk-2, exhibiting IC50 values of 0.016 nM for TYK2-JH2 and 0.31 nM for JAK1-JH2. This compound is valuable for investigating the role of Tyk-2 in inflammatory and autoimmune diseases. Its high potency and selectivity make it a critical tool in the research of signaling pathways associated with these conditions. -
JAK Inhibitor
GDC-9918 is a selective Janus kinase (JAK) inhibitor that modulates JAK signaling pathways, critical for cytokine and growth factor signaling. It demonstrates potent inhibition of JAK activity, making it a valuable tool in investigating immune response and inflammatory processes. This compound is applicable in various research settings, including studies on autoimmune diseases, hematological disorders, and potential oncology applications. -
JAK3/BTK Inhibitor
JAK3/BTK-IN-4 is a potent inhibitor targeting both JAK3 and BTK kinases, which are critical in the modulation of immune responses involved in autoimmune diseases. By simultaneously inhibiting the BTK/JAK3 signaling pathway, JAK3/BTK-IN-4 demonstrates synergistic effects, making it a valuable tool for studying JAK3 and BTK-related pathologies. This compound is particularly relevant for research applications focused on therapeutic strategies for autoimmune disorders. -
JAK1 Inhibitor
JAK1-IN-10 is a selective inhibitor of Janus kinase 1 (JAK1), characterized by its cyano-substituted cyclic hydrazine structure. This compound exhibits potent inhibition of JAK1 activity, making it a valuable tool for investigating the role of JAK1 in various cellular signaling pathways. Its application is particularly relevant in research areas such as immunology, oncology, and inflammatory diseases, where modulation of JAK1 can influence disease progression and therapeutic outcomes. -
TYK2/JAK2 Inhibitor
JAK2/TYK2-IN-1 is a selective inhibitor targeting TYK2 and JAK2, demonstrating IC50 values of 9 nM for TYK2 and 157 nM for JAK2. This compound exhibits notable anti-inflammatory activity, making it valuable for research into inflammatory diseases and immune responses. Its specificity for TYK2 highlights its potential in understanding signal transduction pathways and therapeutic interventions in related conditions. -
JAK3 Inhibitor
JAK3-IN-14 is a selective JAK3 inhibitor that demonstrates potent activity with an IC50 of 38 nM for JAK3 and 600 nM for JAK2. This compound effectively inhibits IL-4 and IL-3 induced proliferation of TF-1 cells, exhibiting IC50 values of 600 nM and 500 nM, respectively. JAK3-IN-14 is valuable for research applications related to cytokine signaling and immune response modulation. -
Tyk2 Inhibitor
Tyk2-IN-18 is a potent inhibitor of tyrosine kinase 2 (Tyk2), effectively blocking its activity. This compound demonstrates a strong inhibitory effect on JAK2-JH2, with an IC50 value of less than 10 nM. Tyk2-IN-18 is valuable for research applications focused on autoimmune diseases, inflammatory disorders, and cancer, facilitating the investigation of Tyk2 signaling pathways. -
SYK/JAK Inhibitor
SYK/JAK-IN-1 is a dual inhibitor targeting SYK and JAK2, exhibiting IC50 values of less than 5 nM for both kinases. This compound demonstrates significant anti-inflammatory and anti-proliferative activities, making it a valuable tool for research involving hematological malignancies and autoimmune disorders. Its potent inhibition profile allows for the exploration of signaling pathways associated with SYK and JAK2, facilitating studies in cancer biology and immunology. -
JAK1 Inhibitor
MMT3-72 is a selective inhibitor of Janus kinase 1 (JAK1), demonstrating effective modulation of the JAK-STAT signaling pathway. This compound significantly reduces phosphorylated STAT3 (p-STAT3) levels in models of dextran sulfate sodium (DSS)-induced colitis, highlighting its potential role in inflammatory bowel disease research. MMT3-72 serves as a valuable tool for studying JAK1-related signaling mechanisms and therapeutic interventions in related conditions. -
JAK1 Inhibitor
JAK1-IN-17 is a highly selective inhibitor of Janus kinase 1 (JAK1), exhibiting a Ki of 1.9 nM. This compound maintains effective potency in whole blood due to its low whole blood shift, making it a valuable tool for hematological studies. Additionally, JAK1-IN-17 is a nitrile-containing analogue that displays weak reversible inhibition of CYP3A4, demonstrated by an IC50 of 7.9 μM. Researchers can utilize JAK1-IN-17 in cancer research to explore the therapeutic potential of JAK1 inhibition in various malignancies. -
JAK3 Inhibitor
JAK3-IN-12 is a potent inhibitor of Janus kinase 3 (JAK3), exhibiting IC50 values of 9.5 nM, 18 nM, and 42 nM for JAK3, JAK1, and JAK2, respectively. This compound serves as a valuable tool for investigating the role of JAK3 in various biological processes, particularly in the context of autoimmune diseases such as rheumatoid arthritis. Its selective inhibition can facilitate understanding of JAK3's function and its potential as a therapeutic target in related research applications. -
JAK inhibitor
JAK kinase-IN-1 is a selective inhibitor of Janus kinase (JAK) family members, effectively targeting TYK2, JAK1, JAK2, and JAK3 with IC50 values of 4.2 nM, 32 nM, 27 nM, and 3473 nM, respectively. This compound demonstrates significant biological activity in modulating JAK-mediated signaling pathways, making it a valuable reagent for research on immune responses and inflammatory diseases. Its specificity and potency contribute to its utility in investigating the role of JAK kinases in various cellular processes and therapeutic applications. -
JAK1 Inhibitor
JAK1-IN-19 is a potent inhibitor of Janus kinase 1 (JAK1), demonstrating IC50 values of 0.02 nM for JAK1, 0.5 nM for JAK2, 91 nM for JAK3, and 0.2 nM for TYK2. This compound exhibits enhanced intrinsic clearance in both rat and human models. JAK1-IN-19 is applicable for research in atopic dermatitis and other autoimmune diseases, facilitating the investigation of JAK1 signaling pathways and their role in inflammatory responses. -
JAK1/2/3 Inhibitor
INCB16562 is a selective inhibitor targeting JAK1 and JAK2, with a notable preference for JAK1 over JAK3. It effectively inhibits interleukin-6 (IL-6)-induced phosphorylation of STAT3, thereby blocking the proliferation and survival of myeloma cells reliant on IL-6 for growth. Furthermore, INCB16562 demonstrates antitumor activity in vivo by reducing the growth of myeloma xenografts in murine models. This compound shows potential for advancing research in multiple myeloma therapies. -
JAK1/2 Inhibitor
JAK1/2-IN-2 is a highly selective inhibitor of the Janus kinase 1 and 2 (JAK1/2) pathways, demonstrating Ki values of 2 nM and 0.6 nM, respectively. This compound is instrumental in research focused on the modulation of cytokine signaling pathways and holds potential for therapeutic applications in autoimmune diseases and hematological malignancies. Its potent inhibition of JAK1/2 makes it a valuable tool for understanding the role of these kinases in various biological processes. -
JAK2 Inhibitor
Tkip is a selective inhibitor of JAK2, targeting the JAK2 autophosphorylation site. It effectively inhibits JAK2 autophosphorylation and the phosphorylation of the IFN-γ receptor subunit IFNGR-1, thereby reducing the antiviral effects of IFN-γ and downregulating MHC Class I molecule expression. Tkip is a valuable tool for investigating the IFN-γ signaling pathway and its implications in various biological processes. -
BET/JAK2/FLT3 Inhibitor
SG3-179 is a selective inhibitor of BET bromodomain proteins, with additional activity against JAK2 and FLT3. This compound effectively reduces HOXB13 protein expression, demonstrating potential relevance in the study of multiple myeloma (MM1.S). SG3-179 is a valuable tool for research involving epigenetic regulation and signaling pathways associated with hematological malignancies. -
JAK3/BTK Inhibitor
JAK3/BTK-IN-1 is a potent dual inhibitor targeting JAK3 and BTK, key proteins implicated in autoimmune diseases. By simultaneously blocking the BTK/JAK3 signaling pathway, this compound demonstrates synergistic effects that could enhance therapeutic outcomes. JAK3/BTK-IN-1 is suitable for research into JAK3 kinase and BTK-related diseases, facilitating the exploration of innovative treatment strategies in immunology and related fields. -
JAK Inhibitor
Tyk2-IN-17 is a selective inhibitor of the Janus kinase 2 (TYK2). This compound effectively impedes the activity of TYK2, which is crucial in various signaling pathways associated with immune regulation and inflammation. Tyk2-IN-17 is primarily used in research focusing on autoimmune diseases, inflammatory disorders, and cancer biology, providing insights into therapeutic strategies for conditions mediated by aberrant JAK signaling. -
JAK Inhibitor
PF-1367550 is a pan-JAK inhibitor that selectively targets Janus kinase enzymes. It is demonstrated to reduce the release of pro-inflammatory cytokines CXCL9, CXCL10, and CXCL11 from primary airway epithelial cells. This compound is valuable for research in inflammatory diseases and the modulation of immune responses. -
JAK Inhibitor
CEE321 is a potent pan-JAK inhibitor that exhibits an IC50 value of 54 nM. It effectively inhibits key biomarkers associated with atopic dermatitis, making it a valuable tool for research in inflammatory skin conditions and related therapeutic studies. Its broad activity against various JAK isoforms facilitates investigations into the signaling pathways involved in immune responses. -
Aurora Kinase A/JAK2 Inhibitor
AJI-100 is a dual-target inhibitor that selectively inhibits Aurora kinase A and JAK2 with IC50 values of 12.7 nM and 18.5 nM, respectively. By directly blocking Aurora kinase A, AJI-100 disrupts T cell mitosis and cell polarity, while its inhibitory effect on JAK2 activation prevents STAT3 phosphorylation. This compound is valuable for research focused on modulating immune responses and has potential applications in the prevention of graft-versus-host disease (GVHD). -
JAK2-STAT5 Activator
Methionyl-methionine (Met-Met) functions as a JAK2-STAT5 activator, enhancing intracellular substrate availability. This compound has been shown to significantly promote the expression of α-s1 casein (αS1-CN) in mammary explants, mediated through the activation of JAK2-STAT5 and mTOR signaling pathways. Its role in modulating these critical pathways makes it a valuable tool for research in lactation biology and protein synthesis studies. -
JAK3 Inhibitor
CP-690550A is a selective inhibitor targeting Janus kinase 3 (JAK3), with notable efficacy against JAK2 as well. This compound exhibits significant immunosuppressive properties and is primarily utilized in research focused on autoimmune diseases and transplant rejection. Its ability to modulate cytokine signaling pathways makes it a valuable tool for studying immune response mechanisms. -
JAK2 Inhibitor
NMS-P953 is a potent orally active inhibitor of JAK2, exhibiting an IC50 of 0.008 μM. This compound demonstrates significant antitumor activity, making it a valuable tool for cancer research. It is particularly useful in studies focusing on JAK2-related signaling pathways and therapeutic applications in hematological malignancies. -
JAK2 Inhibitor
BVB808 is a selective JAK2 inhibitor, exhibiting approximately 10-fold selectivity for JAK2 over other JAK family members in vitro. This compound effectively inhibits JAK2 activity, leading to a reduction in STAT5 phosphorylation, which in turn disrupts JAK2-dependent cell proliferation and survival signaling pathways. BVB808 is utilized in cancer research, particularly in studies focusing on malignancies driven by JAK2 signaling dysregulation. -
JAK3/Syk Inhibitor
R-348 choline is a potent, orally active inhibitor of Janus kinase 3 (JAK3) and spleen tyrosine kinase (Syk). This compound effectively reduces the expression levels of pro-inflammatory cytokines, including interferon-gamma (IFN-γ), interleukin-6 (IL-6), and interleukin-10 (IL-10). R-348 choline is primarily utilized in research related to acute cardiac allograft rejection and other autoimmune conditions where JAK3 and Syk signaling play critical roles. -
JAK3 Inhibitor
JAK3-IN-19 is a selective inhibitor of Janus kinase 3 (JAK3), a critical component in cytokine signaling pathways. Inhibition of JAK3 has been shown to affect the proliferation and survival of cancer cells. This compound is valuable for research applications focused on understanding the role of JAK3 in various malignancies and exploring its potential as a therapeutic target in cancer treatment.
