Epigenetics
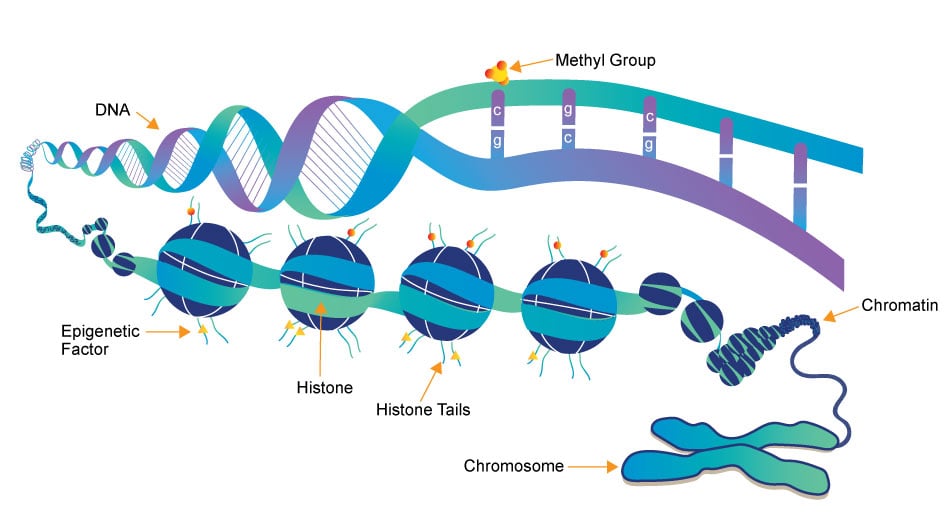
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
EZH2 Inhibitor
DM-01 is a potent and selective inhibitor of EZH2, a key regulator of histone methylation. It demonstrates significant biological activity in the context of diffuse large B-cell lymphoma (DLBCL), follicular lymphoma (FL), and genetically defined solid tumors associated with SNF5/INI-1/SMARCB1 alterations. This compound facilitates research into the molecular mechanisms driving these malignancies and offers potential therapeutic insights for targeted cancer treatment. -
PRMT5 Inhibitor
PRMT5-IN-39 is a potent inhibitor of protein arginine methyltransferase 5 (PRMT5), targeting the enzymatic activity involved in arginine methylation. This compound exhibits significant anti-cancer properties, making it a valuable tool for studying the role of PRMT5 in tumorigenesis and cellular signaling pathways. Researchers can utilize PRMT5-IN-39 to investigate its therapeutic potential in various cancer models and assess its effects on cell growth and proliferation. -
PRMT7 Inhibitor
PRMT7-IN-2 is a selective inhibitor of Protein Arginine Methyltransferase 7 (PRMT7) with an IC50 value of 0.50 μM. This compound effectively arrests the cell cycle at the G0/G1 phase and promotes apoptosis, leading to inhibition of cell growth both in vivo and in vitro. PRMT7-IN-2 decreases monomethylarginine levels associated with PRMT7 activity and modulates epithelial and mesenchymal marker expression, enhancing the levels of E-cadherin while reducing those of N-cadherin, Vimentin, and ZEB2. Such properties make it a valuable tool for studying cancer biology and cellular transformation mechanisms. -
HIF-1α/EZH2 Inhibitor
DYB-03 is an orally active inhibitor of HIF-1α and EZH2. It effectively suppresses migration, invasion, and angiogenesis in lung cancer cells as well as human umbilical vein endothelial cells (HUVECs), demonstrated both in vitro and in vivo. Additionally, DYB-03 induces apoptosis in A549 and H460 cells that are resistant to 2-ME2 and GSK126, highlighting its potential in overcoming resistance in lung cancer therapies. -
MLL1-WDR5 Interaction Inhibitor
HBI-2375 is a selective inhibitor of the MLL1-WDR5 interaction, exhibiting a strong binding affinity with an IC50 of 4.48 nM. This compound effectively inhibits proliferation in MV4;11 leukemia cells, with an IC50 of 3.17 µM. Additionally, HBI-2375 disrupts histone methyltransferase activity, making it a valuable tool for research on leukemia, glioma, and glioblastoma. Its ability to penetrate the blood-brain barrier and its oral bioavailability further enhance its potential for in vivo studies. -
PRMT6 Allosteric Inhibitor
SGC6870 is a selective allosteric inhibitor of the protein arginine methyltransferase 6 (PRMT6), exhibiting an IC50 of 77 nM. This compound functions by modulating PRMT6 activity, thereby influencing cellular processes such as gene expression and signal transduction. SGC6870 is useful for research focused on epigenetic regulation and the role of PRMTs in various diseases, including cancer and neurodegenerative disorders. -
Histone Methyltransferase Inhibitor
RL5a is an inhibitor of the histone methyltransferase SETD8. It effectively disrupts the methylation of lysine 27 on histone H3, playing a crucial role in the regulation of gene expression. This compound is utilized in research focused on epigenetics and the modulation of cellular processes, making it a valuable tool for studies on cancer biology and developmental processes. -
PRMT5 Inhibitor
(R)-AMG-193 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5), functioning as a methionine adenosine transferase (MTA)-cooperative inhibitor. This compound demonstrates significant antitumor activity, particularly in MTAP-deficient tumor cells, with an IC50 value of 0.107 μM. It effectively inhibits PRMT5, allowing for the preferential targeting of tumor cells while sparing normal cells with intact MTAP. (R)-AMG-193 is a valuable tool for research into cancer biology and therapeutic strategies involving PRMT5 inhibition. -
EZH2 Inhibitor
EPZ011989 trifluoroacetate is a selective inhibitor of the enzyme EZH2, exhibiting a Ki value of less than 3 nM. This compound is notable for its ability to effectively inhibit histone methylation, thereby demonstrating significant anti-tumor activity. EPZ011989 trifluoroacetate is applicable in cancer research, providing insights into the role of EZH2 in tumorigenesis. Additionally, it features an alkyne group that facilitates click chemistry applications, allowing for copper-catalyzed azide-alkyne cycloaddition (CuAAc) with compatible azide-containing molecules. -
EZH1/2 Inhibitor
(S)-HH2853 is a potent dual inhibitor of EZH1 and EZH2, specifically targeting the EZH2_Y641F variant with an IC50 of less than 100 nM. This pyridine-based compound demonstrates significant biological activity, making it a valuable tool for research in anti-tumor and autoimmune disease pathways. Its ability to modulate histone methylation presents opportunities for exploring epigenetic regulation in various therapeutic contexts. -
PRMT5 Inhibitor
PRMT5-IN-1 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5), exhibiting an IC50 of 11 nM for the PRMT5/MEP50 complex. This hemiaminal compound can be metabolized to aldehydes, allowing it to form covalent adducts with C449 under physiological conditions. PRMT5-IN-1 is valuable for investigating the role of PRMT5 in various biological processes, including gene regulation and oncogenesis, making it a useful tool for cancer research and other studies involving protein methylation. -
ASH1L Inhibitor
AS-99 free base is a potent and selective inhibitor of the ASH1L histone methyltransferase, with an IC50 of 0.79 µM and a dissociation constant (Kd) of 0.89 µM. It demonstrates significant anti-leukemic activity by inhibiting cell proliferation, inducing apoptosis and differentiation, and downregulating MLL fusion target genes. AS-99 effectively reduces leukemia burden in vivo, making it a valuable tool for research into targeted therapies for leukemia and other ASH1L-related malignancies. -
Histone Methyltransferase Inhibitor
EML741 is a selective inhibitor of the histone methyltransferases G9a and GLP, characterized by an IC50 value of 23 nM for G9a and a Kd of 1.13 μM. Additionally, EML741 inhibits DNMT1 with an IC50 of 3.1 μM while demonstrating no significant effect on DNMT3a or DNMT3b. This compound exhibits low cytotoxicity, is membrane permeable, and is capable of crossing the blood-brain barrier, making it a valuable tool for epigenetic research and potential therapeutic applications. -
EZH2 Inhibitor
EPZ011989 hydrochloride is a potent inhibitor of the Zeste Homolog 2 (EZH2) enzyme, demonstrating a Ki value of less than 3 nM. This compound exhibits notable anti-tumor activity, making it a valuable tool in cancer research. Additionally, EPZ011989 serves as a click chemistry reagent, featuring an alkyne group that enables copper-catalyzed azide-alkyne cycloaddition (CuAAc) with azide-containing molecules, facilitating various biochemical applications. -
Histone Methyltransferase Inhibitor
Igermetostat is a potent inhibitor of histone methyltransferase EZH2, which plays a crucial role in gene silencing and epigenetic regulation. This compound exhibits significant activity in both in vivo and in vitro models, making it valuable for cancer research. Its application in studies of tumor biology and potential therapeutic strategies underscores its importance in exploring epigenetic modulation in various malignancies. -
DOT1L Inhibitor
S-N6-Methyladenosylhomocysteine is a potent inhibitor of the histone methyltransferase DOT1L, exhibiting an IC50 value of 0.29 μM. By selectively targeting DOT1L, this compound plays a crucial role in cancer research, particularly in studies investigating the epigenetic regulation and its implications in oncogenesis. It serves as a valuable tool for understanding the biochemical pathways associated with methylation-driven cancers and may facilitate the development of therapeutic strategies. -
WDR5 Degrader
MS33 is a potent WDR5 degrader that functions through E3 ligase VHL-mediated degradation. It exhibits binding affinities of 870 nM for VCB and 120 nM for WDR5, effectively targeting this protein for proteasomal degradation. MS33 is primarily utilized in research related to acute myeloid leukemia, aiding in the understanding of WDR5's role in cancer biology. -
EZH2 Inhibitor
EZH2-IN-14 is a highly selective inhibitor of EZH2, a histone methyltransferase, with an IC50 of 12 nM. By specifically targeting and inhibiting the methyltransferase activity of EZH2 within the PRC2 complex, EZH2-IN-14 effectively reduces levels of H3K27me3. This compound exhibits over 200-fold selectivity for EZH2 compared to the closely related EZH1, making it a valuable tool for research into epigenetic regulation and cancer biology. -
SETD2 Inhibitor
EZM0414 TFA is a potent and selective inhibitor of SETD2, exhibiting an IC50 of 18 nM in biochemical assays and 34 nM in cellular assays. This orally active reagent is suitable for investigating its effects on relapsed or refractory multiple myeloma and diffuse large B-cell lymphoma. EZM0414 TFA facilitates valuable research into the role of SETD2 in cancer biology and therapeutic development. -
SETD7 Inhibitor
PFI-2 hydrochloride is a potent and selective inhibitor of SET domain containing lysine methyltransferase 7 (SETD7). It exhibits significant inhibitory activity with an IC50 value of 2.0 nM, making it an essential tool for studying the role of SETD7 in various biological processes. PFI-2 hydrochloride is valuable for investigating mechanisms underlying chronic kidney disease and inflammation, particularly in the context of renal fibrosis research. -
SETDB1-TTD Inhibitor
SETDB1-TTD-IN-1 is a selective inhibitor targeting the tandem Tudor domain of the SET domain bifurcated protein 1 (SETDB1-TTD), exhibiting a binding affinity (Kd) of 88 nM. This compound enhances the methyltransferase activity of SETDB1, making it a valuable tool for investigating the biological roles and disease associations of SETDB1-TTD in epigenetic regulation and cellular processes. Researchers can utilize SETDB1-TTD-IN-1 to explore its implications in various biological contexts. -
PRMT5 Inhibitor
PF-06939999 is a potent, orally active inhibitor of protein arginine methyltransferase 5 (PRMT5), functioning as a S-adenosylmethionine (SAM) competitive antagonist. It effectively suppresses the expression of symmetric dimethylarginine (SDMA) protein, with an IC50 value of 1.1 nM in A427 cells. Due to its ability to inhibit PRMT5 activity, PF-06939999 demonstrates significant antitumor effects, making it valuable for cancer research focused on epigenetic regulation and methylation pathways. -
JMJD1C Inhibitor
JMJD1C-IN-1 is a selective inhibitor of JMJD1C with an IC50 of 0.59 μM and a Kd of 1.96 μM. This compound effectively disrupts the binding of JMJD1C to the H3K9me2 peptide substrate as demonstrated in the HTRF assay, showing an IC50 of 1.47 μM. JMJD1C-IN-1 enhances tumor immunotherapy research by impairing intratumoral regulatory T (Treg) cell fitness through the accumulation of H3K9me2, which downregulates PD1 expression, and by reducing STAT3 demethylation, thereby promoting STAT3 activation. Furthermore, it exhibits dose-dependent antitumor efficacy across various mouse cancer models, including fibrosarcoma, melanoma, lung cancer, hepatocellular carcinoma, and colorectal cancer. -
PRMT1 Inhibitor
Furamidine dihydrochloride is a selective inhibitor of protein arginine methyltransferase 1 (PRMT1), exhibiting an IC50 of 9.4 μM. This compound demonstrates significant selectivity for PRMT1 over PRMT5, PRMT6, and PRMT4, making it an essential tool for studying PRMT-related biological processes. In addition, Furamidine dihydrochloride inhibits tyrosyl-DNA phosphodiesterase 1 (TDP-1) competitively and reversibly, showing enhanced activity with duplex DNA substrates. Furthermore, it possesses antiparasitic properties, expanding its potential applications in chemical and biological research. -
SETD2 Inhibitor
EPZ-719 is a selective inhibitor of the SETD2 enzyme, exhibiting an IC50 value of 0.005 μM. This compound demonstrates significant anticancer activity, making it a valuable tool for research in cancer biology and therapeutic development. EPZ-719 may be utilized to explore the role of SETD2 in tumorigenesis and to evaluate potential treatment strategies targeting this pathway. -
DOT1L Inhibitor
Dot1L-IN-4 is a potent inhibitor of the DOT1L enzyme, exhibiting an IC50 of 0.11 nM. It effectively disrupts the activity of the telomeric silencing 1-like protein, making it a valuable tool for research into epigenetic regulation and gene expression modulation. Its application extends to studies on leukemia and other cancer types where DOT1L is implicated, facilitating investigations into therapeutic approaches targeting this pathway. -
HDAC Inhibitor
HDAC-IN-48 is a potent inhibitor of histone deacetylases (HDACs) that exhibits significant cytotoxicity, with a GI50 of approximately 20 nM. This hybrid molecule incorporates pharmacophores from SAHA and CETZOLE, effectively inducing ferroptosis while inhibiting HDAC activity. Additionally, HDAC-IN-48 features an alkyne group, allowing it to participate in copper-catalyzed azide-alkyne cycloaddition (CuAAc) reactions, making it a valuable tool for click chemistry applications in chemical biology and therapeutic research. -
HDAC11 Inhibitor
HDAC11-IN-3 is a selective inhibitor of HDAC11, exhibiting an IC50 of 4.1 nM. This compound demonstrates potent anti-acute myeloid leukemia (AML) activity against U937 and OCI-AML2 cell lines with an IC50 of 10 μM. It effectively induces apoptosis, cell cycle arrest, and differentiation while upregulating iron transporters transferrin (TF) and transferrin receptor (TFRC). Additionally, HDAC11-IN-3 activates the p62-Keap1-Nrf2-HMOX1 pathway, resulting in elevated intracellular iron levels and subsequent ferroptosis in AML cells. This reagent is suited for studies investigating the molecular mechanisms of AML and can be utilized alone or in combination with other therapeutic agents like Cytarabine. -
LSD1 Inhibitor
Higenamine hydrochloride is a selective inhibitor of LSD1, with an IC50 value of 1.47 μM. This compound exhibits anti-inflammatory and antibacterial properties, and has been shown to attenuate IL-1β-induced apoptosis via the ROS-mediated PI3K/Akt signaling pathway. Additionally, Higenamine hydrochloride protects brain cells from oxygen deprivation and promotes bone formation in osteoporosis through the SMAD2/3 pathway. Its versatile applications make it suitable for research in cancer, inflammation, cardiorenal syndrome, and related diseases. -
LSD1 Inhibitor
GSK-LSD1 is a selective inhibitor of lysine-specific demethylase 1 (LSD1). This compound demonstrates significant biological activity by reducing food intake and body weight, while also enhancing insulin sensitivity and glycemic control in mouse models of obesity. Additionally, GSK-LSD1 shows promise in alleviating non-alcoholic fatty liver disease (NAFLD) and inhibiting cytokine release triggered by SARS-CoV-2 in peripheral blood mononuclear cells from COVID-19 patients. Furthermore, GSK-LSD1 has been identified as a potential therapeutic agent for suppressing cancer growth and metastasis. -
JAK3 Inhibitor
DPPY is a potent JAK3 inhibitor with an IC50 of less than 10 nM, demonstrating high selectivity for protein tyrosine kinases including EGFR and BTK. It exhibits significant anti-proliferative activity against B-cell lymphoma cells. DPPY holds potential for research applications related to idiopathic pulmonary fibrosis (IPF) and other conditions involving aberrant JAK3 signaling. -
JAK Inhibitor
MJ04 is a selective inhibitor targeting Janus Kinase 3 (JAK 3) with an IC50 of 2.03 nM. This compound effectively inhibits T cell differentiation and reduces pro-inflammatory cytokine production in lipopolysaccharide-induced macrophages. Additionally, MJ04 exhibits favorable pharmacokinetic properties in mice and has demonstrated the ability to promote hair growth in a dihydrotestosterone-induced model of androgenetic alopecia (AGA), showing minimal toxicity (LD50 > 2 g/kg). -
PARP-1 Inhibitor
CEP-6800 is a potent inhibitor of PARP-1, known for its ability to enhance the efficacy of chemotherapeutic agents. It effectively reduces poly(ADP-ribose) accumulation induced by irinotecan and temozolomide in LoVo and HT29 xenograft models. Additionally, CEP-6800 demonstrates potential in suppressing tumor growth in Calu-6. This compound is valuable for research in cancer biology and therapy development. -
PARP
PARP-1/2-IN-1 is a highly effective inhibitor of the poly(ADP-ribose) polymerases PARP-1 and PARP-2, exhibiting IC50 values of 0.51 nM and 23.11 nM, respectively. This compound plays a critical role in cancer research by targeting DNA repair mechanisms, enhancing the efficacy of chemotherapeutic agents, and potentially improving cancer treatment outcomes. It is a valuable tool for studying PARP-related cellular processes and investigating therapeutic strategies in oncology. -
PARP-1/-2 inhibitor
CEP-9722 is a selective, orally active inhibitor of PARP-1 and PARP-2, exhibiting IC50 values of 20 nM and 6 nM, respectively. This compound demonstrates significant anticancer activity, making it a valuable tool for research in cancer therapy and DNA repair mechanisms. Its ability to inhibit these critical enzymes positions CEP-9722 as an important reagent for studying cellular responses to DNA damage and tumor susceptibility to therapeutic agents. -
PARP7 Inhibitor
PARP7-IN-21 is a potent inhibitor of PARP7, demonstrating an IC50 of less than 10 nM. This compound effectively interferes with the activity of PARP7, which is involved in the regulation of cellular processes such as DNA repair and signaling pathways related to stress response. PARP7-IN-21 is valuable for research applications focused on cancer biology, neurodegenerative diseases, and other conditions associated with PARP7 dysregulation. -
CDK9/PARP Inhibitor
CDK9/PARP-IN-1 is a potent inhibitor of CDK9 and PARP1, demonstrating IC50 values of 118 nM and 107 nM, respectively. This dual inhibition results in significant antiproliferative effects across various cancer cell lines, making it a valuable tool for cancer research. CDK9/PARP-IN-1 is particularly relevant for studies investigating the therapeutic potential of targeting these pathways in oncology. -
PARP1/CDK12 Inhibitor
Antitumor agent-104 is a potent inhibitor of PARP1 and CDK12, targeting critical pathways in DNA damage repair in tumors. By inhibiting PARP1 enzymatic activity, it effectively reduces PAR protein levels, thus impairing the cellular mechanisms that protect tumor cells. This compound serves as a valuable tool in cancer research, especially in studies focused on understanding tumor biology and exploring novel therapeutic strategies. -
PARP1 Inhibitor
PARP1-IN-53 is a potent PARP1 inhibitor with an IC50 of 0.1 nM, demonstrating high selectivity over PARP2, which has an IC50 of 23 nM. This quinazolinone derivative effectively interferes with the poly(ADP-ribose) polymerase 1 enzyme, making it a valuable compound for cancer research. Its specific action on PARP1 enables detailed studies into the mechanisms of DNA repair and cell survival in oncology. -
PARP PROTAC Degrader
PROTAC PARP1 degrader-2 is a targeted protein degradation compound designed to specifically degrade PARP1. With a DC50 of less than 10 nM in MDA-MB-231 cells, it demonstrates potent efficacy in inducing degradation. Additionally, this compound inhibits cell viability in MDA-MB-436 cells with an IC50 of less than 100 nM, making it a valuable tool for research in cancer therapeutics and the mechanistic study of PARP1 function. -
PARP Inhibitor
INO-1001 mesylate is a selective inhibitor of poly (ADP-ribose) polymerase (PARP), a critical enzyme involved in DNA repair processes. It enhances the sensitivity of cancer cells to radiation therapy by disrupting DNA repair mechanisms, leading to increased necrotic cell death. This compound is of interest in cancer research, particularly in studies aimed at overcoming resistance to radiation and improving therapeutic outcomes in tumorigenesis. -
PARP-1/2 Inhibitor
CEP-8983 is a potent inhibitor of PARP-1 and PARP-2, with IC50 values of 20 nM and 6 nM, respectively. This compound effectively enhances the sensitivity of chemotherapy-resistant cell lines and subcutaneous xenograft models to the anticancer agents Temozolomide and Camptothecin. Its ability to disrupt DNA repair mechanisms makes CEP-8983 a valuable tool for cancer research, particularly in studies focusing on therapeutic resistance and combination therapies. -
PARP-1 Inhibitor
ST7710AA1 is a potent inhibitor of PARP-1, exhibiting an IC50 value of 0.07 µM. This compound demonstrates significant antiproliferative and anticancer activity, making it a valuable tool for research in oncology and cellular biology. Its ability to inhibit PARP-1 can be leveraged to explore mechanisms of cancer cell survival and the effects of DNA damage repair pathways. -
PARP-1 Inhibitor
8-Chloroquinazolin-4-ol is a potent inhibitor of the PARP-1 enzyme, exhibiting an IC50 value of 5.65 μM. This compound serves as a nicotinamide mimic and plays a significant role in research focused on DNA repair mechanisms and cancer therapies. Its ability to modulate PARP-1 activity makes it a valuable tool for exploring therapeutic strategies in various disease models. -
PARP-1 Inhibitor
Benzo[c][1,8]naphthyridin-6(5H)-one is a potent inhibitor of poly(ADP-ribose) polymerase-1 (PARP-1) and aurora kinase A, exhibiting IC50 values of 0.311 μM and 5.5 μM, respectively. This compound demonstrates low micromolar affinity for human adenosine receptors AR A1 and hA2A, with Ki values of 4.6 and 4.8 μM. Due to its mechanistic action, Benzo[c][1,8]naphthyridin-6(5H)-one is valuable for research applications targeting DNA repair pathways and cancer therapies. -
PARP-1 Inhibitor
BSI-401 is an orally active inhibitor of PARP-1, a key enzyme involved in DNA repair processes. This compound demonstrates significant anti-cancer activity, particularly in pancreatic cancer, both as a monotherapy and in combination with Oxaliplatin. BSI-401 is valuable for research into therapeutic strategies targeting DNA damage response pathways in cancer treatment. -
PARP-2 Inhibitor
UPF-1035 is a selective inhibitor of PARP-2, exhibiting an IC50 value of 0.15 μM. This compound has been shown to increase CA1 pyramidal cell loss in the hippocampus, indicating its role in neuroprotection. UPF-1035 can be utilized in research focused on neurodegenerative diseases and the mechanisms of neuronal cell survival. -
PARP1 Inhibitor
PARP1-IN-19 is a potent inhibitor of poly (ADP-ribose) polymerase 1 (PARP1), a key enzyme involved in DNA repair mechanisms. This compound exhibits significant antitumor activity, making it a valuable tool in cancer research and therapeutic development. It is primarily utilized in studies focusing on the modulation of DNA damage response pathways and the exploration of combination therapies in oncology. -
PARP Inhibitor
NU1064 dihydrochloride is a selective inhibitor of poly(ADP-ribose) polymerase (PARP), an enzyme involved in DNA repair mechanisms. This compound enhances the cytotoxic effects of DNA-methylating agents, such as MTIC, in a concentration-dependent manner. It is valuable for research in cancer biology, particularly in studies focusing on enhancing the efficacy of chemotherapeutic agents and understanding mechanisms of DNA repair inhibition. -
PARP Inhibitor
KU-0058684 is a selective PARP inhibitor, exhibiting an IC50 of 3.2 nM for PARP-1. This reagent effectively impairs DNA double strand break repair, making it a valuable tool for investigating DNA damage response mechanisms. Its application extends to studying the therapeutic potential of PARP inhibition in various cancer models.
