-
GPR40 activator
GPR40 Activator 2 is a potent GPR40 activator from patents WO 2012147516 A1, WO 2012046869A1 and WO 2011078371 A1. -
GPR88 receptor agonist
(1R,2R)-2-PCCA hydrochloride is a diastereomer of 2-PCCA, and acts as a potent GPR88 receptor agonist, with an EC50 of 3 nM in cell-free assay, and 603 nM in cell assay. -
SARD ligand
(R)-UT-155 (compound 11) is a selective androgen receptor degrader (SARD) ligand. Less active than the S-isomer. -
estrogen receptor (ERα) antagonist
AZD9496 maleate is a potent and selective estrogen receptor (ERα) antagonist with IC50 of 0.28 nM. AZD9496 maleate is an orally bioavailable selective oestrogen receptor degrader (SERD). -
thyroid hormone receptor agonist
Eprotirome is a liver-selective thyroid hormone receptor agonist. -
GPR40 agonist
AMG 837 sodium salt is a potent GPR40 agonist(EC50=13 nM) with a superior pharmacokinetic profile and robust glucose-dependent stimulation of insulin secretion in rodents. -
progesterone receptor agonist
Nomegestrol acetate is a potent, highly selective progestogen, which is characterized as a full agonist at the progesterone receptor, with no or minimal binding to other steroid receptors, including the androgen and glucocorticoid receptors. -
oestrogen receptor antagonist
Enclomiphene citrate is a potent and orally active oestrogen receptor antagonist, with antioestrogenic property. -
D prostanoid receptor 2 antagonist
Timapiprant sodium (OC000459 sodium) is a potent, selective, and orally active D prostanoid receptor 2 (DP2, also known as CRTH2) antagonist. -
direct renin inhibitor
Aliskiren D6 Hydrochloride (CGP 60536 D6 Hydrochloride) is is deuterium labeled Aliskiren, which is a direct renin inhibitor with IC50 of 1.5 nM. -
glucocorticoid receptor agonist
Mapracorat is a novel non-steroidal selective glucocorticoid receptor agonist. -
estrogen receptor (ER) inhibitor
WAY-169916 is a pathway-selective inhibitor of estrogen receptor (ER) that acts by inhibiting NF-kB transcriptional activity. -
antiestrogenic properties
FLTX1 is a fluorescent Tamoxifen derivative that can specifically label intracellular Tamoxifen-binding sites (estrogen receptors) under permeabilized and non-permeabilized conditions. FLTX1 exhibits the potent antiestrogenic properties of Tamoxifen in breast cancer cells. FLTX1 is devoid of the estrogenic agonistic effect on the uterus. -
cell permeable TgPrxII inhibitor
Conoidin A is a cell permeable inhibitor of T. gondii enzyme peroxiredoxin II (TgPrxII). -
ERRα agonist
Cholesterol is also an endogenous estrogen-related receptor α (ERRα) agonist. -
5α-reductase inhibitor
Finasteride Carboxaldehyde is a metabolite of Finasteride in human bile (M2 metabolite) that can be used for proteomics research. -
Antiandrogenic agent
4'-Methoxyresveratrol (4'-O-Methylresveratrol) is a polyphenol derived from Dipterocarpaceae, with antiandrogenic, antifungal and anti-inflammatory activities. -
synthetic glucocorticoid receptor agonist
Methylprednisolone (NSC-19987, Medrol) acetate is a synthetic glucocorticoid receptor agonist, used to achieve prompt suppression of inflammation. -
GPR109A agonist
Monomethyl fumarate, an active metabolite of Dimethyl fumarate (DMF), is a potent GPR109A agonist. Monomethyl fumarate has the potential for multiple neuroprotective pathways and other models of retinal disease. -
Estrogen receptor degrader
SAR439859 is an orally available, nonsteroidal selective estrogen receptor degrader/downregulator (SERD). -
Androgen receptor antagonist
TRC253, also known as JNJ63576253, is a potent and orally active androgen receptor antagonist. -
GnRH peptide agonist
Triptorelin is a synthetic gonadotropin-releasing hormone (GnRH) peptide agonist that binds to the GnRH receptor. It inhibits the growth of DU145, LNCaP, and PC3 prostate and OVCAR-3 ovarian cancer cells. -
GnRH receptor antagonist
Cetrorelix diacetate (SB-075 diacetate) is a potent gonadotropin-releasing hormone (GnRH) receptor antagonist with an IC50 of 1.21 nM. - (D-Trp12,Tyr34)-pTH (7-34) amide (bovine) is a potent and competitive antagonist of parathyroid hormone (PTH), with a Ki of 69 nM in bovine renal cortical membrane. (D-Trp12,Tyr34)-pTH (7-34) amide (bovine) can be used for growth and development regulation.
- ELA-21 (human) is an apelin receptor agonist with a pKi of 8.52. ELA-21 (human) completely inhibits Forskolin-induced cAMP production and stimulates β-arrestin recruitment with subnanomolar potencies. ELA-21 (human) is an agonist in G-protein-dependent and -independent pathways.
- (Glu2)-TRH, a metabolically stable analogue of Thyrotropin-releasing hormone (TRH), is a negative modulator for the cholinergic effect of TRH in the mouse brain. (Glu2)-TRH significantly attenuates TRH-induced hippocampal extracellular acetylcholine release. (Glu2)-TRH is not metabolized by thyroliberinase. (Glu2)-TRH manifests neuroprotective, antidepressant, anticonvulsant in the CNS.
-
GnRH Receptor agonist
Histrelin acetate, a GnRH analogue, is a GnRH Receptor agonist. Histrelin acetate increases serum luteinising hormone (LH), follicle stimulating hormone (FSH) and testosterone levels. Histrelin acetate can be used in the research of prostate cancer, endometriosis. -
SLU-PP-332 is a pan-Estrogen Receptor/ERR agonist with EC50 values of 98, 230 and 430 nM for ERRα, ERRβ and ERRγ, respectively. SLUPP-332 enhances mitochondrial function and cellular respiration in skeletal muscle cell lines. SLU-PP-332 has the potential to study metabolic diseases as well as improve muscle function.
-
PROTAC
Luxdegalutamide (ARV-766) is an orally active proteolysis-targeting chimera (PROTAC) designed to selectively degrade the androgen receptor (AR), including clinically relevant resistance-associated mutants such as T878A, H875Y, and L702H. By inducing AR ubiquitination and proteasomal degradation, Luxdegalutamide effectively suppresses AR signaling and exhibits potent antitumor activity. It is a promising therapeutic agent and research tool for studying castration-resistant prostate cancer (CRPC) and mechanisms of AR-driven oncogenesis. - Prednisolone hemisuccinate is a synthetic glucocorticoid derivative of cortisol, commonly used in the treatment of inflammatory and autoimmune disorders. It exerts anti-inflammatory and immunosuppressive effects by modulating gene expression through glucocorticoid receptor activation.
-
THR-β agonist
ALG-055009 is a selective thyroid hormone receptor β (THR-β) agonist with an EC50 of 0.063 μM. It effectively reduces total cholesterol levels in high-fat diet-induced rat models and is a promising compound for research related to metabolic dysfunction-associated fatty liver disease (MAFLD). -
GPR35 agonist
Pamoic acid disodium is a potent agonist of GPR35, with an EC50 of 79 nM. It induces GPR35 internalization and activates ERK1/2 signaling with EC50 values of 22 nM and 65 nM, respectively. Additionally, it effectively recruits β-arrestin2 to GPR35 and exhibits antinociceptive properties, supporting its potential in pain research. -
GPR55 antagonist
ML192 is a selective antagonist of G protein-coupled receptor 55 (GPR55), effectively inhibiting GPR55-mediated signaling pathways. It blocks β-arrestin trafficking, suppresses ERK1/2 phosphorylation, and prevents PKCβII translocation, thereby interfering with downstream cellular responses. ML192 is a valuable tool for studying the physiological and pathological roles of GPR55 in processes such as inflammation, pain, cancer, and metabolic regulation. -
GPR35 agonist
Pamoic acid (Embonic acid) is a potent agonist of G protein-coupled receptor 35 (GPR35), with an EC₅₀ of 79 nM. Activation of GPR35 by pamoic acid is associated with both neuroprotective and anti-inflammatory effects, making it a valuable compound for research into neurological disorders and inflammatory diseases. Its pharmacological profile suggests potential therapeutic applications in conditions where GPR35-mediated signaling plays a regulatory role. -
PROTAC AR/AR-V7 degrader
PROTAC AR/AR-V7 Degrader-1 (27c) is a PROTAC-based dual degrader targeting both full-length androgen receptor (AR) and its splice variant AR-V7, which is implicated in resistance to androgen deprivation therapies. It exhibits DC₅₀ values of 2.67 μM for AR and 2.64 μM for AR-V7, effectively promoting their proteasomal degradation. By eliminating both isoforms, compound 27c induces apoptosis in AR-driven cancer cells, making it a promising therapeutic candidate for castration-resistant prostate cancer (CRPC) and other AR/AR-V7–dependent malignancies. -
PROTAC AR degrader
ARD-1676 is an orally bioavailable PROTAC degrader of the androgen receptor (AR), composed of an AR-binding ligand and a cereblon-recruiting moiety. It effectively induces AR degradation both in vitro and in vivo, and demonstrates significant antitumor activity by inhibiting VCaP prostate cancer xenograft growth in mouse models. ARD-1676 represents a promising therapeutic strategy for targeting AR-driven malignancies.
Endocrinology-Hormones
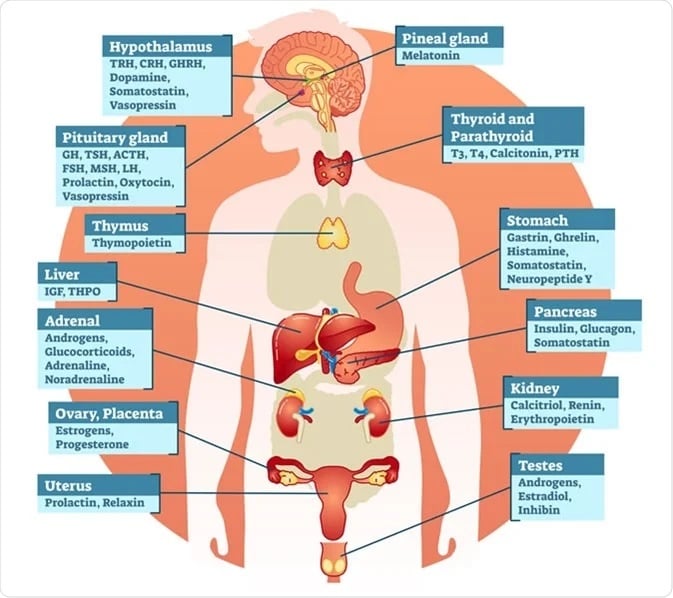
Small molecules play a pivotal role in Endocrinology Research. These are low molecular weight compounds that have a significant impact on the endocrine system, hormones, and their receptors. Here are some key aspects of how small molecules are involved in this field:
- Hormone Mimetics and Inhibitors: Small molecules are used to develop synthetic compounds that mimic the actions of hormones or inhibit their effects. For example, drugs like metformin for diabetes management and selective estrogen receptor modulators (SERMs) for breast cancer treatment are used to either mimic or block hormonal activity.
- Receptor Modulation: Small molecules can bind to hormone receptors and modulate their activity. This is crucial in developing drugs that target specific hormone receptors, like the use of small molecule agonists and antagonists to regulate thyroid hormone receptors.
- Metabolism Regulation: Endocrinology research often focuses on metabolism and how hormones like insulin regulate it. Small molecules are employed to understand and develop drugs targeting enzymes involved in metabolism, such as glucagon-like peptide-1 (GLP-1) agonists for diabetes treatment.
- Steroid Hormone Production: Small molecules may be utilized to influence the production of steroid hormones in the adrenal glands or gonads. This is essential for conditions like Cushing's syndrome or polycystic ovary syndrome (PCOS).
- Hormone Assays: In laboratory research, small molecules are used as tracers or markers in hormone assays. For instance, small molecule fluorophores can be attached to antibodies to detect hormone levels in blood samples.
Drug Development: Endocrinology research relies on small molecules as potential drug candidates. Researchers design and test small molecules for their effectiveness in modulating hormonal pathways, with the goal of developing new therapies for endocrine disorders.
In summary, small molecules are indispensable tools in Endocrinology Research, enabling scientists to better understand the endocrine system's intricacies and develop novel treatments for a wide range of hormonal disorders and conditions. Their versatility and specificity make them valuable assets in advancing our knowledge of endocrinology and improving patient care.
Endocrinology Disease Products
Endocrinology Research Products
Kisspeptin Receptor
Leptin Receptors
Melanocortin (MC) Receptors
Mineralocorticoid Receptors
Galanin Receptors
TRH Receptors
Ghrelin Receptors
Natriuretic Peptide Receptors
NPY Receptors
Motilin Receptor
PTH Receptor
