Epigenetics
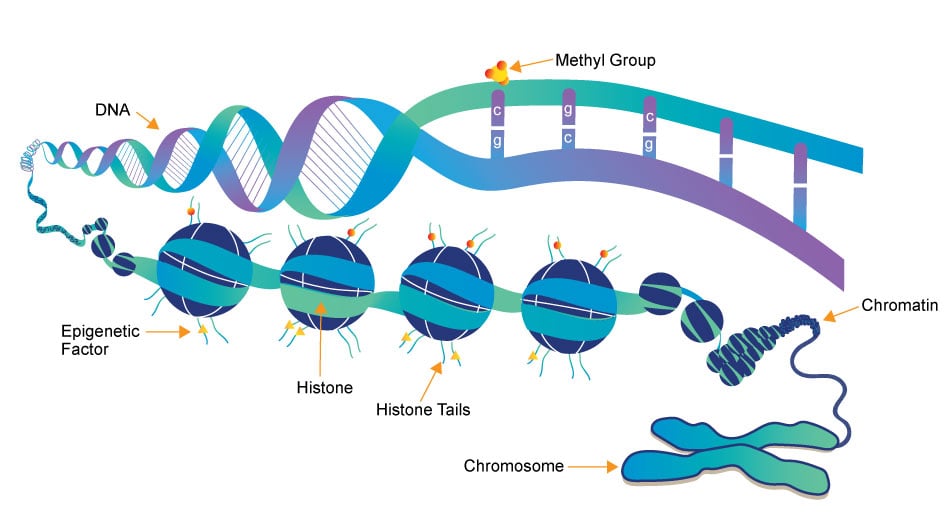
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
PRMT5-MTA complex inhibitor
MRTX9768 is a highly potent, selective, and orally bioavailable first-in-class inhibitor of the PRMT5-MTA complex, demonstrating targeted inhibition of protein arginine methyltransferase 5 (PRMT5) in MTAP-deleted tumors with potential antineoplastic activity. -
SMYD3 inhibitor
EM127 (compound 11c) is a highly selective, covalent SMYD3 inhibitor with a KD of 13 μM and site-specific binding. It potently inhibits ERK1/2 phosphorylation, suppresses transcriptional regulation of SMYD3 target genes, and provides sustained inhibition of methyltransferase activity. EM127 is suitable for cancer research, particularly in SMYD3-positive tumors. -
PRMT4 inhibitor
EPZ0025654 (GSK3536023) is a potent inhibitor of protein arginine methyltransferase 4 (PRMT4, also known as CARM1), with an IC50 of 3 nM. -
DNMT inhibitor
MC3343 is a DNMT inhibitor (EC50: 5.7 μM) and potent DNA binder that inhibits tumor proliferation by arresting osteosarcoma (OS) cells in the G1 or G2/M phases. It promotes osteoblastic differentiation by specifically reactivating genes involved in this physiological process. -
NSUN2 inhibitor
MY-1B is a covalent inhibitor of the RNA methyltransferase NSUN2 (IC50: 1.3 μM), stereoselectively targeting active-site cysteine residues (C271). It also covalently binds to PSME1, disrupting the proteasome regulatory complex and downregulating specific MHC-I subtype presentation. -
5-Methyldeoxycytidine
5-Methyl-2'-deoxycytidine (5mdC) is an endogenous substrate of DNA methyltransferases, including mammalian 5-C-MTase, and binds to DNA in a manner dependent on DNA stem-loop structure formation. It directs de novo DNA methylation by serving as a methylation mark, promoting methylation of adjacent CpG sites in single-stranded DNA via cis-acting mechanisms. 5mdC regulates DNA methylation patterns by recruiting methyltransferases to specific chromatin regions, influencing chromatin condensation and gene expression. In plant cells, its distribution correlates with cell proliferation and differentiation stages, with lower methylation levels in proliferating cells and higher levels in differentiated cells. -
TET2 inhibitor
TFMB-(S)-2-HG is a potent inhibitor of TET2 and EglN prolyl hydroxylases. It downregulates Wnt3a and intranuclear β-catenin protein expression, and inhibits osteogenic differentiation of cells. TFMB-(S)-2-HG shows potential for research in acute myeloid leukemia (AML). -
HDAC1/DNA Methyltransferase Inhibitor
Psammaplin A, a marine-derived metabolite, is a potent inhibitor of HDAC1 (IC50: 45 nM), DNA methyltransferases (IC50: 18.6 nM), and aminopeptidase N (IC50: 18 μM). It also suppresses DNA topoisomerase and farnesyl protein transferase activities. As a PPARγ activator, Psammaplin A induces apoptosis and exhibits antitumor, anti-inflammatory, and anti-angiogenic properties. Additionally, it demonstrates antibacterial activity against Gram-positive bacteria by inhibiting DNA synthesis and DNA -
PRMT5/7 inhibitor
DS-437 is a dual inhibitor of PRMT5 and PRMT7, with IC₅₀ values of 6 μM for both enzymes. It exhibits high selectivity over 29 other human methyltransferases, including protein, DNA, and RNA methyltransferases. DS-437 functions as a S-adenosylmethionine (SAM)-competitive inhibitor of PRMT5 and also inhibits DNMT3A and DNMT3B with IC₅₀ values of 52 μM and 62 μM, respectively. Additionally, DS-437 inhibits the methylation of FOXP3, making it a useful tool for studying epigenetic regulation and immune modulation. -
PRMT5 inhibitor
PRT543 (PRMT5-IN-35) is a potent, selective, and orally active inhibitor of protein arginine methyltransferase 5 (PRMT5), with an IC₅₀ value of 1 nM. It effectively targets PRMT5-mediated epigenetic regulation and demonstrates strong potential for use in cancer research, particularly in malignancies driven by dysregulated arginine methylation. -
PRMT5 inhibitor
TNG908 is a MTAP (methylthioadenosine phosphorylase)-synergistic inhibitor of PRMT5 (protein arginine methyltransferase 5). It is orally active and capable of crossing the blood–brain barrier, making it suitable for targeting both systemic and central nervous system tumors. TNG908 selectively exploits MTAP deletion—a common alteration in cancers—to enhance its antitumor efficacy, making it a promising agent for cancer research and precision oncology. -
PRMT5/MTA inhibitor
Navlimetostat is a potent, orally active, and selective inhibitor of the PRMT5-MTA complex. It exhibits IC₅₀ values of 3.6 nM for the PRMT5-MTA complex and 20.5 nM for PRMT5 alone. Navlimetostat binds to the PRMT5-MTA complex with exceptionally high affinity (K\_D = 0.14 pM). It demonstrates strong antineoplastic activity both in vitro and in vivo, making it a promising candidate for cancer research and therapeutic development targeting PRMT5-driven malignancies. -
PRMTs inhibitor
AMI-1 free acid is a potent, cell-permeable, and reversible inhibitor of protein arginine N-methyltransferases (PRMTs). It inhibits human PRMT1 and yeast Hmt1p with IC₅₀ values of 8.8 μM and 3.0 μM, respectively. AMI-1 functions by blocking the binding of peptide substrates to PRMTs, making it a useful tool for studying arginine methylation and its role in gene regulation, signal transduction, and disease. -
PRMTs inhibitor
MS023 dihydrochloride is a potent, selective, and cell-permeable inhibitor of type I protein arginine methyltransferases (PRMTs). It exhibits strong inhibitory activity with IC₅₀ values of 30 nM for PRMT1, 119 nM for PRMT3, 83 nM for PRMT4, 4 nM for PRMT6, and 5 nM for PRMT8. MS023 is a valuable tool for studying PRMT-dependent epigenetic regulation and has potential applications in cancer and other PRMT-associated diseases. -
PRMT5 inhibitor
LLY-284 is the diastereomer of LLY-283 and exhibits significantly reduced activity compared to LLY-283. While LLY-283 is a potent inhibitor of protein arginine methyltransferase 5 (PRMT5) with strong anticancer potential, LLY-284 serves primarily as a less active control compound in studies evaluating PRMT5-targeted cancer therapeutics. -
PRMT5 inhibitor
BRD0639 is a first-in-class inhibitor that disrupts the interaction between protein arginine methyltransferase 5 (PRMT5) and its substrate adaptor proteins. It functions as a PRMT5 binding motif (PBM)-competitive agent, selectively interfering with PBM-dependent PRMT5 activities. BRD0639 is a valuable chemical probe for studying PRMT5-mediated methylation and its role in epigenetic regulation and disease. -
EZH1/EZH2 inhibitor
Tulmimetostat (CPI-0209) is an orally active, dual inhibitor of EZH1 and EZH2, key enzymatic components of the polycomb repressive complex 2 (PRC2) involved in histone methylation and gene silencing. By targeting EZH2, Tulmimetostat effectively suppresses aberrant gene repression associated with tumorigenesis. It exhibits antitumor activity and is currently being investigated in studies involving a variety of solid tumors and hematologic malignancies. -
DNMT inhibitor
5-Fluoro-2'-deoxycytidine (FdCyd) is a fluoropyrimidine nucleoside analogue that functions as a DNA methyltransferase (DNMT) inhibitor. It also serves as a tumor-selective prodrug of 5-fluoro-2′-dUMP, a thymidylate synthase inhibitor. FdCyd induces cell cycle arrest in tumor cells by activating the DNA damage response and exhibits potent anti-tumor activity, making it a promising agent in epigenetic and chemotherapeutic cancer research. -
DNMT1 inhibitor
DC-05 is a DNA methyltransferase 1 (DNMT1) inhibitor with an IC₅₀ of 10.3 μM and a binding affinity (K\_d) of 1.09 μM. It interferes with DNMT1-mediated DNA methylation, making it a useful compound for studying epigenetic regulation and exploring potential therapeutic applications in diseases involving aberrant DNA methylation. -
TET inhibitor
Bobcat339 hydrochloride is a potent and selective cytosine-based inhibitor of TET (ten-eleven translocation) enzymes, with IC₅₀ values of 33 μM for TET1 and 73 μM for TET2. By inhibiting TET-mediated DNA demethylation, it modulates epigenetic regulation and gene transcription. Bobcat339 hydrochloride is a valuable tool in epigenetics research and serves as a potential lead compound for developing therapeutics targeting DNA methylation-related diseases. -
DNMT1 inhibitor
GSK-3484862 is a non-covalent and selective inhibitor of DNA methyltransferase 1 (DNMT1). It effectively induces DNA hypomethylation, making it a promising agent for cancer therapy. In murine embryonic stem cells, GSK-3484862 mediates robust and targeted demethylation with minimal off-target toxicity, supporting its potential for epigenetic reprogramming and therapeutic applications. -
DNMT1 inhibitor
GSK-3685032 is a first-in-class, reversible, non-time-dependent, and noncovalent inhibitor that selectively targets DNA methyltransferase 1 (DNMT1), with an IC₅₀ of 0.036 μM. It induces robust DNA demethylation, leading to transcriptional activation and inhibition of cancer cell growth, making it a promising candidate for epigenetic therapy in oncology. -
LSD1 inhibitor
Bomedemstat (IMG-7289) is an orally active, irreversible inhibitor of lysine-specific demethylase 1 (LSD1). By inhibiting LSD1, it increases methylation of histone marks H3K4 and H3K9, leading to altered gene expression. Bomedemstat exhibits potent anti-cancer activity by inhibiting cancer cell proliferation and inducing apoptosis, and is being explored as a therapeutic agent in hematologic malignancies and other cancers. -
LSD1 inhibitor
Pulrodemstat benzenesulfonate (CC-90011 benzenesulfonate) is a potent, selective, reversible, and orally active inhibitor of lysine-specific demethylase 1 (LSD1), with an IC₅₀ of 0.25 nM. It exhibits significantly lower inhibitory activity against related enzymes such as LSD2, MAO-A, and MAO-B. CC-90011 induces differentiation in acute myeloid leukemia (AML) and small cell lung cancer (SCLC) cells and demonstrates strong anticancer activity, making it a promising candidate for epigenetic cancer therapy. -
Menin-MLL Interaction Inhibitor
Ziftomenib (KO-539) is an orally active inhibitor of the menin–MLL (KMT2A) interaction, designed to disrupt oncogenic gene expression in MLL-rearranged cancers. It exhibits potent antitumor activity and is under investigation for the treatment of acute leukemias driven by MLL rearrangements or NPM1 mutations. Ziftomenib corresponds to compound 151 in patent WO2017161028A1. -
Menin-KMT2A inhibitor
Bleximenib (JNJ-75276617) is an orally active and highly selective menin–KMT2A (MLL) interaction inhibitor, with IC50 values of 0.1 nM in humans, 0.045 nM in mice, and ≤0.066 nM in dogs. It effectively inhibits the proliferation of tumor cells and induces apoptosis and differentiation, particularly in malignancies driven by KMT2A rearrangements. Bleximenib is a promising therapeutic candidate for the study and treatment of leukemia and other menin-dependent cancers. -
Menin-MLL Interaction Inhibitor
DSP5336 is an orally active small molecule inhibitor that targets the interaction between menin and MLL (mixed-lineage leukemia) proteins. By disrupting this protein–protein interaction, DSP5336 impairs leukemogenic gene expression programs driven by MLL rearrangements. It is currently undergoing clinical evaluation for the treatment of patients with relapsed or refractory acute leukemia, particularly those harboring MLL rearrangements. -
Menin-KMT2A inhibitor
Bleximenib (JNJ-75276617) oxalate is an orally active and highly selective inhibitor of the menin–KMT2A (MLL) interaction, with IC50 values of 0.1 nM in humans, 0.045 nM in mice, and ≤0.066 nM in dogs. It effectively inhibits tumor cell proliferation and induces apoptosis and differentiation, particularly in cancers driven by KMT2A rearrangements. Bleximenib oxalate is a promising candidate for research in leukemia and other menin–KMT2A-dependent malignancies. -
G9a inhibitor
RK-701 is a highly selective and non-genotoxic inhibitor of G9a histone methyltransferase, with an IC₅₀ of 23–27 nM. It selectively upregulates fetal hemoglobin (HbF), γ-globin, and *BGLT3* expression, while downregulating H3K9me2 levels. RK-701 also exhibits inhibitory effects on the transcriptional repressors BCL11A and ZBTB7A, making it a promising candidate for the treatment of hemoglobinopathies such as sickle cell disease and β-thalassemia. -
NSD2 degrader
UNC8153 is a potent and selective degrader targeting nuclear receptor-binding SET domain-containing protein 2 (NSD2), with a binding affinity (Kd) of 24 nM. It induces proteasome-dependent degradation of NSD2 by incorporating a simple warhead, leading to a reduction in both NSD2 protein levels and the associated H3K36me2 chromatin mark in cells. UNC8153 is a valuable chemical tool for investigating NSD2 function and epigenetic regulation. -
NSD antagonist
UNC6934 is a potent and selective chemical probe targeting the N-terminal PWWP1 domain of NSD2 (nuclear receptor-binding SET domain protein 2). It binds NSD2-PWWP1 with a Kd of 80 nM (measured by SPR) and exhibits high selectivity over 14 other PWWP domains, including NSD3-PWWP1. In U2OS cells, UNC6934 disrupts the interaction between NSD2-PWWP1 and H3K36me2-marked nucleosomes, with an IC₅₀ of 1.09 μM as determined by NanoBRET assay. It is a valuable tool for studying NSD2 chromatin interactions and epigenetic regulation. -
MAT2A inhibitor
AG-270 is a first-in-class, reversible, and orally active methionine adenosyltransferase 2A (MAT2A) inhibitor. It functions as an allosteric and noncompetitive inhibitor, with an IC₅₀ of 14 nM. AG-270 is being explored for its therapeutic potential in cancers with MTAP deletions, where MAT2A inhibition leads to selective antitumor effects via disruption of methylation-dependent processes. -
MAT2A inhibitor
IDE397 (GSK-4362676) is a potent and selective inhibitor of methionine adenosyltransferase 2A (MAT2A). It exhibits strong anti-tumor activity, particularly in patient-derived xenograft (PDX) models with MTAP (methylthioadenosine phosphorylase) deletion. By targeting MAT2A in MTAP-deficient tumors, IDE397 exploits a synthetic lethality approach, making it a promising candidate for precision oncology research. -
PRMT4 inhibitor
TP-064 is a potent and selective inhibitor of protein arginine methyltransferase 4 (PRMT4/CARM1), with an IC50 of less than 10 nM. It effectively inhibits the dimethylation of BAF155 and MED12 with IC50 values of 340 nM and 43 nM, respectively. TP-064 shows minimal activity against other PRMT family members, except for PRMT6 (IC50 = 1.3 μM). It exhibits anticancer activity, making it a valuable tool for studying PRMT4-related epigenetic regulation and cancer therapy. -
DK/PI3K/BRD4 Inhibitor
SRX3177 is a potent triple inhibitor targeting CDK4/6, PI3K, and BRD4, with IC50 values of <2.5 nM for CDK4, 3.3 nM for CDK6, 79 nM for PI3Kα, 83 nM for PI3Kδ, 3.18 μM for PI3Kγ, and 33 nM and 89 nM for BRD4 BD1 and BD2, respectively. It exhibits broad cytotoxic activity against cancer cells while sparing normal epithelial cells, highlighting its potential as a targeted cancer therapeutic with reduced toxicity. - 6-Demethoxytangeretin is a flavonoid compound isolated from *Citrus reticulata* with demonstrated anti-inflammatory and anti-allergic properties. It inhibits IL-6 production and the expression of related genes in human mast cells by modulating the ALK and MAPK signaling pathways. Additionally, 6-Demethoxytangeretin enhances CRE-mediated transcription in hippocampal neurons, indicating potential neuroregulatory effects.
-
PRMT5 inhibitor
AM-9747 (PRMT5-IN-25) is a highly potent PRMT5 inhibitor with a Ki value of 0.06 nM. It exhibits strong antiproliferative activity, making it a valuable candidate for research in PRMT5-driven cancers and epigenetic regulation. -
JAK2/FLT3 inhibitor
Flonoltinib is a potent and orally active dual JAK2/FLT3 inhibitor with IC50 values of 0.7 nM for JAK2, 4 nM for FLT3, 26 nM for JAK1, and 39 nM for JAK3. It exhibits strong anti-cancer activity and is a promising candidate for the treatment of hematologic malignancies and other JAK/FLT3-driven cancers. -
SHP2 inhibitor
Migoprotafib (GDC-1971; compound 199) is a selective SHP2 inhibitor that suppresses the MAPK/ERK signaling pathway. It exhibits antitumor activity and is under investigation for its potential in targeting SHP2-driven cancers. -
Aurora A/B inhibitor
Tinengotinib (TT00420) is an orally bioavailable, spectrally selective small-molecule kinase inhibitor targeting Aurora A/B (IC50=1.2–3.3 nM), FGFR1/2/3 (IC50=1.5–3.5 nM), VEGFRs, JAK1/2, and CSF1R. It disrupts Aurora kinase-mediated cell cycle progression, inducing G2/M arrest, inhibits the FGFR/JNK-JUN signaling pathway, and activates the MEK/ERK-dependent apoptotic pathway. Tinengotinib exhibits potent anti-tumor proliferation, pro-apoptotic, anti-angiogenic, and tumor microenvironment-modulating activities. It is a promising candidate for research in triple-negative breast cancer (TNBC), gallbladder cancer, and tumor immune microenvironment studies.
