Epigenetics
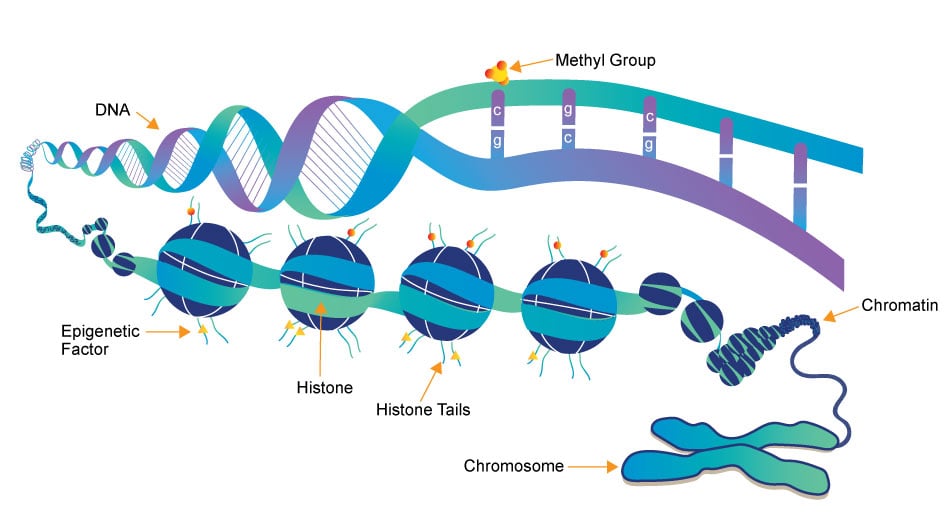
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
PARP Inhibitor
PARP/EZH2-IN-1 is a potent dual inhibitor targeting PARP and EZH2, with respective IC50 values of 6.87 nM and 36.51 nM. This reagent demonstrates significant biological activity in the treatment of triple-negative breast cancer, particularly in cells with wild-type BRCA. Its unique mechanism of action makes it a valuable tool for research into cancer biology and therapeutic development. -
Histone Methyltransferase Inhibitor
PRMT5-IN-55 is a selective inhibitor of the histone methyltransferase PRMT5, demonstrating a pIC50 value of 9.6 at a concentration of 10 nM. This compound plays a crucial role in modulating epigenetic processes by inhibiting arginine methylation, which can influence gene expression and cellular differentiation. PRMT5-IN-55 is valuable for research applications focused on cancer biology, epigenetics, and the therapeutic exploration of PRMT5 in various diseases. -
PRMT1 Inhibitor
PRMT1-IN-3 is a selective inhibitor of protein arginine methyltransferase 1 (PRMT1), demonstrating an IC50 of 4.11 μM. In addition to its primary activity against PRMT1, it also inhibits PRMT6 and PRMT8 with IC50 values of 23.3 and 30.1 μM, respectively. PRMT1-IN-3 effectively reduces asymmetric dimethylarginine (ADMA) levels and histone H4R3me2a modification in triple-negative breast cancer (TNBC) cells, leading to cell cycle arrest, apoptosis, and decreased migration and colony formation in MDA-MB-231 cells. This compound serves as a potential chemotherapeutic sensitizer for Paclitaxel and is valuable for research concerning TNBC. -
GSK-3β/G9a Inhibitor
GSK-3β/G9a-IN-1 is a selective inhibitor of GSK-3β and G9a, acting through competitive mechanisms with IC50 values of 0.8 μM and 1.1 μM, respectively. This compound is effective in lowering tau phosphorylation and reducing Aβ aggregation, making it relevant for Alzheimer's disease research. Additionally, GSK-3β/G9a-IN-1 influences chromatin dynamics by inhibiting H3K9me2 and modulating members of the SAGA complex. Its ability to improve memory and restore social behaviors highlights its potential as a therapeutic agent in neurodegenerative conditions. -
PRMT5 Inhibitor
PRMT5-IN-29 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5), exhibiting an IC50 of 1.5 μM. This compound demonstrates significant biological activity and is suitable for advancing research in cancer biology, particularly in the context of targeted therapies. Its potency and oral bioavailability make it a valuable tool for investigating the role of PRMT5 in various malignancies. -
EZH2 Inhibitor
EZH2-IN-23 is a selective inhibitor of the EZH2 enzyme, exhibiting potent inhibition of the PRC2 complex with an IC50 of 0.8 nM. It effectively reduces H3K27 trimethylation in cellular assays with an IC50 of 40 nM, making it a valuable tool for studying epigenetic regulation. Additionally, EZH2-IN-23 demonstrates favorable pharmacokinetic properties in rat models, featuring 100% oral bioavailability, which enhances its potential for in vivo research applications. -
PRMT5 Inhibitor
PRMT5-IN-43 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5). This compound exhibits significant biological activity by disrupting PRMT5-mediated methylation processes, which play a crucial role in cancer cell proliferation and survival. Its application in cancer research makes it a valuable tool for investigating the mechanisms of oncogenesis and potential therapeutic interventions targeting PRMT5. -
PRMT5•MTA Inhibitor
PRMT5-MTA-IN-5 is an irreversible inhibitor targeting the PRMT5•MTA complex, with an IC50 value of 1.15 nM. This compound effectively inhibits arginine methylation, disrupting ribosomal RNA processing and affecting the expression of proteins associated with the cell cycle. PRMT5-MTA-IN-5 demonstrates potent antiproliferative effects in MTAP-deficient tumor cells, making it a valuable tool for research on MTAP-deficient solid tumors, including liver, breast, and pancreatic cancers. -
SETD8 Inhibitor
[Nle20] H4 peptide (16−23) is a potent inhibitor of the histone methyltransferase SETD8, exhibiting a Kd of 0.14 μM. This peptide competes with histone H4 for binding to the substrate site of SETD8, effectively blocking its methylation activity. As a result, [Nle20] H4 peptide (16−23) serves as a valuable tool in cancer research, offering potential applications in the development of therapeutic strategies targeting epigenetic modulation. -
PRMT5 Inhibitor
PRMT5-IN-44 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5). This compound exhibits significant anti-proliferative effects in various cancer cell lines, making it a valuable tool for studying the role of PRMT5 in tumorigenesis. PRMT5-IN-44 is particularly relevant for research applications focused on cancer biology and therapeutic development targeting PRMT5-mediated pathways. -
LSD1/G9a Inhibitor
LSD1-IN-20 is a potent dual non-covalent inhibitor of lysine-specific demethylase 1 (LSD1) and G9a, exhibiting Ki values of 0.44 and 0.68 μM, respectively. This compound demonstrates significant antiproliferative effects in THP-1 leukemia and MDA-MB-231 breast cancer cell lines, with IC50 values of 0.51 and 1.60 μM over 72 hours. LSD1-IN-20 serves as a valuable tool for research focused on epigenetic regulation and its implications in cancer biology. -
PRMT3 Inhibitor
PRMT3-IN-5 is an allosteric inhibitor of protein arginine methyltransferase 3 (PRMT3) with an IC50 value of 291 nM. This compound is valuable for studying the biological roles of PRMT3 and its implications in various diseases. Its specificity and potency make PRMT3-IN-5 a useful tool in epigenetic research and therapeutic investigations. -
Histone Methyltransferase Inhibitor
PRMT5-IN-9 is an inhibitor specifically targeting protein arginine methyltransferase 5 (PRMT5). This compound exhibits potent inhibitory activity with an IC50 of 0.01 μM, making it a valuable tool for cancer research. PRMT5-IN-9 can be utilized to study the role of arginine methylation in gene expression, cellular signaling, and tumor progression, thereby aiding in the exploration of novel therapeutic strategies. -
G9a Inhibitor
CSV0C018875 hydrochloride is a selective inhibitor of G9a (EHMT2), effectively blocking its enzymatic activity in both enzyme and cell-based assays. This compound demonstrates a lower toxicity profile compared to the known G9a inhibitor BIX-01294, making it a valuable tool for research. CSV0C018875 binds tightly to the active site of G9a, enhancing the compound's inhibitory potency and prolonging its residence time. Its potential for improved ADME (absorption, distribution, metabolism, and excretion) and pharmacodynamic properties positions CSV0C018875 as a promising candidate for further biological studies. -
DOT1L Inhibitor
Dot1L-IN-7 is a potent inhibitor of DOT1L (disruptor of telomeric silencing 1-like protein) with an IC50 of 1.0 μM. This compound selectively induces cytotoxic effects in Mixed Lineage Leukemia (MLL)-AF9 cells while leaving E2A-HLF cells unaffected. It serves as a valuable tool for studying the roles of DOT1L in leukemogenesis and evaluating potential therapeutic strategies for MLL. -
PRMT5 Inhibitor
PRMT5-IN-39-d3 is a deuterated inhibitor targeting protein arginine methyltransferase 5 (PRMT5). This orally bioavailable compound is essential for investigating the role of PRMT5 in various cancer models. Its specific inhibition of PRMT5 provides insights into epigenetic regulation and potential therapeutic applications in oncology research. -
EZH2 Inhibitor
EZH2-IN-22 is a potent inhibitor of EZH2, exhibiting IC50 values of less than 0.00051 µM for the EZH2 (Y641N) and EZH2 (Y641F) mutants, and 0.00052 µM for wild-type EZH2. This compound demonstrates significant antiproliferative activity, making it a valuable tool for research in epigenetic regulation and cancer biology. Its application extends to studies of EZH2-related pathways and therapeutic approaches for tumors exhibiting EZH2 mutations. -
PRMT5 Inhibitor
PRMT5-IN-18 is a highly effective inhibitor of protein arginine methyltransferase 5 (PRMT5), a key enzyme implicated in various cancer pathways. This compound demonstrates strong selectivity and potency, making it a valuable tool for investigating the role of PRMT5 in oncogenesis. Its utility in biochemical assays and cellular studies provides insights into potential therapeutic strategies targeting PRMT5-related diseases. -
HDAC6 Inhibirotr
HDAC6-IN-65 is a selective inhibitor of histone deacetylase 6 (HDAC6) with an IC50 of 0.9 nM, demonstrating additional inhibitory effects on HDAC3 with an IC50 of 39.4 nM. This compound induces the accumulation of acetylated α-tubulin and acetylated histone H3 in Neuro-2a cells, serving as a marker for class I HDAC inhibition. HDAC6-IN-65 provides valuable insights in the study of melanoma and related cancer research applications. -
EZH2 Inhibitor
BBDDL2059 is a selective covalent inhibitor targeting the histone methyltransferase EZH2, demonstrating an IC50 of 1.5 nM against the EZH2-Y641F mutation. This compound effectively inhibits lymphoma cell proliferation at nanomolar concentrations, making it a valuable tool for anticancer research. BBDDL2059 is suitable for investigations into the mechanistic roles of EZH2 in cancer biology and therapeutic development. -
PRMT5 Inhibitor
PRMT5-IN-32 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5). It effectively reduces HCT116 cell proliferation, demonstrating an IC50 value of 0.13 μM. This compound is valuable for research investigating the role of PRMT5 in cancer biology and for assessing the therapeutic potential of PRMT5 inhibition in various malignancies. -
Histone Methyltransferase
Prospasmine hydrochloride is an anticholinergic agent that primarily targets histone methyltransferases, influencing gene expression through epigenetic modification. Its biological activity includes the inhibition of glandular secretions and the relaxation of smooth muscles, making it beneficial in the study of gastrointestinal disorders and smooth muscle spasm relief. Additionally, it demonstrates potential as an anesthetic adjuvant, providing a versatile tool for research in both analgesic and gastrointestinal pharmacology. -
PRMT5 Inhibitor
PRMT5-IN-41 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5), demonstrating potent enzymatic inhibition with oral bioavailability. In addition to its primary mechanism, PRMT5-IN-41 exhibits off-target inhibition of the hERG ion channel, with an IC50 value of 1.36 µM. This compound is valuable for research applications investigating the role of PRMT5 in various biological processes, including cancer progression and gene regulation. -
PRMT1 Inhibitor
DCPT1061 is a selective inhibitor of protein arginine methyltransferase 1 (PRMT1), as well as PRMT6 and PRMT8. It demonstrates significant biological activity with a reduced effect on other PRMTs, such as PRMT3, PRMT4, and PRMT5, as well as other epigenetic enzymes. This compound exhibits antitumor effects, making it a valuable tool for research into cancer biology and epigenetic regulation. -
EZH2 Inhibitor
EZH2-IN-4 is a potent inhibitor of the EZH2 enzyme, exhibiting IC50 values of 0.923 nM for wild type EZH2 and 2.65 nM for mutant EZH2. This compound demonstrates significant anti-cancer activity, making it a valuable tool for research in cancer therapeutics and epigenetic regulation. Its oral bioavailability enhances its potential for in vivo studies targeting EZH2-driven malignancies. -
EZH2/HSP90 Inhibitor
EZH2/HSP90-IN-29 is a dual inhibitor targeting both EZH2 and HSP90, exhibiting IC50 values of 6.29 nM for EZH2 and 60.1 nM for HSP90. This compound enhances the expression of apoptosis and necrosis-related genes, induces M-phase cell cycle arrest, and disrupts the reactive oxygen species catabolism pathway. Additionally, EZH2/HSP90-IN-29 has the capability to cross the blood-brain barrier, making it a valuable tool for research in cancer biology and neurodegenerative diseases. -
PRMT5-MTA Inhibitor
PRMT5-MTA-IN-7 is a selective inhibitor of the PRMT5-MTA complex, demonstrating a KD of 236 nM for PRMT5-MTA and 2.84 μM for PRMT5-SAM. It exhibits IC50 values of 4.08 μM and 13.6 μM for these targets, respectively. This compound selectively inhibits the proliferation of MTAP-deficient cancer cells, making it a valuable tool for research in colon cancer and related studies. -
EZH2/EZH1 Inhibitor
EZH2-IN-1 is a selective inhibitor targeting EZH2 and EZH1 through a SAM-competitive mechanism. It demonstrates potent inhibition with IC50 values of 32 nM for wild-type EZH2, 197 nM for the EZH2 Y641N mutant, and 213 nM for EZH1. This compound effectively reduces levels of bulk H3K27me3 and H3K27me2, highlighting its potential utility in research related to diffuse large B cell lymphoma. -
PRMT5 Inhibitor
PRMT5-IN-40 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5). It exhibits potent inhibition of PRMT5 enzymatic activity, which is critical for regulating various biological processes, including gene expression, cell proliferation, and apoptosis. This compound is valuable for researchers studying epigenetic modifications and the role of PRMT5 in cancer and other diseases. -
PRMT1 Inhibitor
Furamidine is a selective inhibitor of protein arginine methyltransferase 1 (PRMT1), exhibiting an IC50 of 9.4 μM, while maintaining a considerably lower affinity for PRMT5, PRMT6, and PRMT4. Additionally, Furamidine acts as a reversible, competitive inhibitor of tyrosyl-DNA phosphodiesterase 1 (TDP-1), demonstrating enhanced inhibition with duplex DNA substrates. This compound has applications in research focusing on methylation patterns and DNA repair mechanisms, as well as serving as an antiparasitic agent. -
EZH2 Inhibitor
EZH2-IN-16 is a potent inhibitor of the histone methyltransferase EZH2, targeting both the wild-type and the Y641F mutant forms with IC50 values of 37.6 nM and 79.1 nM, respectively. This compound effectively inhibits the proliferation of WSU-DLCL2 cells, demonstrating a GI50 of 0.2 μM. EZH2-IN-16 is valuable for research applications focused on epigenetic regulation and oncology, particularly in the context of EZH2-related pathways in cancer progression. -
PRMT5 Inhibitor
PRMT5-IN-36 is a potent inhibitor of protein arginine methyltransferase 5 (PRMT5), which plays a critical role in regulating gene expression and cellular signaling. This compound demonstrates significant anticancer activity, making it a valuable tool in cancer research. Its oral bioavailability facilitates in vivo studies, allowing for deeper exploration of PRMT5's involvement in tumorigenesis and potential therapeutic applications in malignancies. -
PRMT5 Inhibitor
PRMT5-IN-34 is a selective inhibitor of protein arginine methyltransferase 5 (PRMT5), which plays a critical role in gene expression and cellular signaling through the methylation of arginine residues on target proteins. This compound demonstrates significant biological activity by modulating PRMT5 activity, thereby influencing various cellular processes including cell proliferation and differentiation. PRMT5-IN-34 is invaluable for research applications aimed at understanding the role of arginine methylation in cancer and other diseases related to dysregulation of methyltransferases. -
EZH2/EED Interaction Inhibitor
SAH-EZH2 is an EZH2/EED interaction inhibitor designed to disrupt the binding between embryonic ectoderm development (EED) and the EZH1/EZH2 complex. This stable α-helical peptide selectively inhibits the trimethylation of histone H3 at lysine 27 (H3K27me3), providing a valuable tool for studying epigenetic regulation. SAH-EZH2 is applicable in research focused on developmental biology and the mechanisms of cancer progression related to the regulation of gene expression by histone modifications. -
EZH2 Inhibitor
Tanshindiol C is an EZH2 inhibitor that competes with S-adenosylmethionine, demonstrating an IC50 of 0.55 μM for inhibiting methyltransferase activity. It activates Nrf2 and Sirtuin 1 (Sirt1) in macrophages, suggesting potential roles in oxidative stress response. Tanshindiol C exhibits anti-cancer properties and may serve as a valuable tool in research related to atherosclerosis and epigenetic regulation. -
Histone Methyltransferase Inhibitor
PRMT5-IN-15 is a potent inhibitor of the histone methyltransferase PRMT5, exhibiting an IC50 value of 0.84 nM. This compound selectively targets PRMT5, disrupting its enzymatic activity and consequently influencing epigenetic regulation. PRMT5-IN-15 is valuable for research into gene expression, cellular differentiation, and cancer biology, particularly in studying the role of arginine methylation in various pathological conditions. -
PRMT5/MTA Complex Inhibitor
(S)-Navlimetostat is a selective inhibitor of the PRMT5/MTA complex, demonstrating an IC50 value of 7070 nM. This compound exhibits significant potential in modulating arginine methylation processes, making it a valuable tool for studying the role of PRMT5 in various biological pathways. It is particularly relevant for research applications in cancer, neurodegenerative diseases, and other conditions associated with dysregulated methylation. -
Histone Methyltransferase Inhibitor
CPI-1328 is a potent inhibitor of the histone methyltransferase EZH2, exhibiting a Ki value of 63 fM. This compound is primarily utilized in research to investigate the role of EZH2 in gene regulation, cancer progression, and epigenetic modifications. It is particularly relevant for studies exploring the therapeutic potential of targeting histone methylation pathways in various malignancies. -
G9a Inhibitor
DCG066 is a selective inhibitor of the lysine methyltransferase G9a, effectively binding to G9a and obstructing its methyltransferase activity in vitro. This compound has been shown to decrease di-methylation levels of histone H3 at lysine 9 (H3K9Me2), leading to inhibition of cell proliferation and induction of apoptosis. Additionally, DCG066 exhibits low cytotoxicity in leukemia cell lines characterized by elevated G9a expression, such as K562, making it a valuable tool for studying the role of G9a in oncogenesis and potential therapeutic applications. -
Histone Methyltransferase
HLCL-61 is a potent small-molecule inhibitor of protein arginine methyltransferase 5 (PRMT5), a key enzyme involved in histone methylation. By selectively targeting PRMT5, HLCL-61 disrupts its activity, leading to alterations in gene expression and potential therapeutic effects in various cancers. This compound serves as a valuable tool for researching the role of arginine methylation in epigenetic regulation and the mechanistic pathways associated with PRMT5 in cancer biology. -
EZH2 Inhibitor
EZH2-IN-17 is a potent EZH2 inhibitor with an IC50 value of 0.95 nM, demonstrating significant selectivity and oral bioavailability. This compound exhibits robust anti-proliferative activity against various lymphoma cell lines, including WSU-DLCL2, Pfeiffer, and Karpas-422, with IC50 values of 2.36 nM, 1.73 nM, and 1.82 nM, respectively. EZH2-IN-17 is useful for research focused on epigenetic modulation and the investigation of EZH2's role in cancer progression. -
PRMT Inhibitor
MS023 trihydrochloride is a potent and selective inhibitor of human type I protein arginine methyltransferases (PRMTs), demonstrating impressive IC50 values of 30 nM for PRMT1, 119 nM for PRMT3, 83 nM for PRMT4, 4 nM for PRMT6, and 5 nM for PRMT8. Its ability to effectively inhibit these enzymes makes it a valuable tool for research into the roles of PRMTs in various biological processes and disease states. This compound is suitable for studies involving cellular signaling, epigenetics, and post-translational modifications. -
EZH2 Inhibitor
(R)-HH2853 is a selective inhibitor of the EZH2 enzyme, specifically targeting the mutant EZH2-Y641F variant with an IC50 of less than 100 nM. This compound demonstrates potential in therapeutic applications related to cancer and autoimmune diseases, enabling researchers to investigate its effects on histone methylation and gene regulation. Its specificity for mutant EZH2 may provide insights into mechanisms of malignancy and contribute to the development of novel treatment strategies. -
PRMT4/PRMT6 Inhibitor
MS049 dihydrochloride is a selective inhibitor of protein arginine methyltransferases PRMT4 and PRMT6, demonstrating IC50 values of 34 nM and 43 nM, respectively. This compound effectively reduces the levels of dimethylated Med12 and H3R2 in HEK293 cells, making it valuable for studying the role of arginine methylation in cellular processes. MS049 dihydrochloride shows low toxicity and does not adversely affect cell proliferation, offering a reliable tool for research into epigenetic regulation and associated pathways. -
G9a Inhibitor
G9a-IN-1 is a targeted inhibitor of the G9a protein (EHMT2), a nuclear histone lysine methyltransferase responsible for the dimethylation of histone H3 at lysine 9 (H3K9me2), a modification linked to transcriptional repression. By inhibiting G9a, G9a-IN-1 plays a crucial role in unraveling the mechanisms of gene silencing associated with various pathological conditions. This compound is valuable for research in autoimmune disorders and cancer, offering insights into therapeutic strategies for these diseases. -
Histone Methyltransferase Inhibitor
PRT543 is a selective inhibitor of the histone methyltransferase PRMT5, targeting key epigenetic modifications associated with various cancers and hematological disorders. This compound demonstrates significant efficacy in inhibiting PRMT5 activity, leading to alterations in gene expression and cellular phenotypes relevant to cancer progression and disorders such as sickle cell disease and hereditary persistence of fetal hemoglobin (HPFH). PRT543 serves as a valuable tool for researchers investigating the role of PRMT5 in epigenetic regulation and its potential as a therapeutic target. -
PRMT6 Degrader
SKLB-0124 is a selective degrader of Protein Arginine Methyltransferase 6 (PRMT6) with an IC50 of 1.6 μM. This compound induces proteasome-dependent degradation of PRMT6, resulting in significant inhibition of cell proliferation and the induction of apoptosis and cell cycle arrest. SKLB-0124 has demonstrated DC50 values of 15.4 μM in HCC827 cells and 16.4 μM in MDA-MB-435 cells, making it a valuable reagent for researching lung and breast cancer. It does not degrade PRMT1 or PRMT8, ensuring specificity in studies involving PRMT6. -
Type I PRMT Inhibitor
GSK3368715 trihydrochloride is a highly selective inhibitor targeting type I protein arginine methyltransferases (PRMTs), demonstrating a remarkable capacity to inhibit PRMT1 (IC50 = 3.1 nM) and other isoforms such as PRMT6 and PRMT8. As an uncompetitive inhibitor of S-adenosyl-L-methionine, this compound significantly affects arginine methylation patterns and alters exon usage. GSK3368715 trihydrochloride has shown promising anti-cancer activity, making it a valuable tool for research into the role of PRMTs in disease mechanisms and therapeutic development. -
PRMT5 Inhibitor
PRMT5-IN-4 is a potent inhibitor of protein arginine methyltransferase 5 (PRMT5), which plays a critical role in tumorigenesis. By blocking PRMT5 activity, this compound exhibits significant anti-tumor effects, making it a valuable tool in cancer research. PRMT5-IN-4 can be utilized in studies investigating the molecular mechanisms of cancer cell proliferation and survival, as well as potential therapeutic strategies targeting PRMT5 in various malignancies. -
DOT1L Inhibitor
Dot1L-IN-6 is a potent inhibitor of the DOT1L enzyme, demonstrating an IC50 value of 0.19 nM. This compound effectively disrupts the function of the telomeric silencing 1-like protein, making it a valuable tool for exploring the role of DOT1L in various biological processes. Researchers can utilize Dot1L-IN-6 in studies related to epigenetics, gene regulation, and the development of therapies for diseases associated with abnormal DOT1L activity.
