Epigenetics
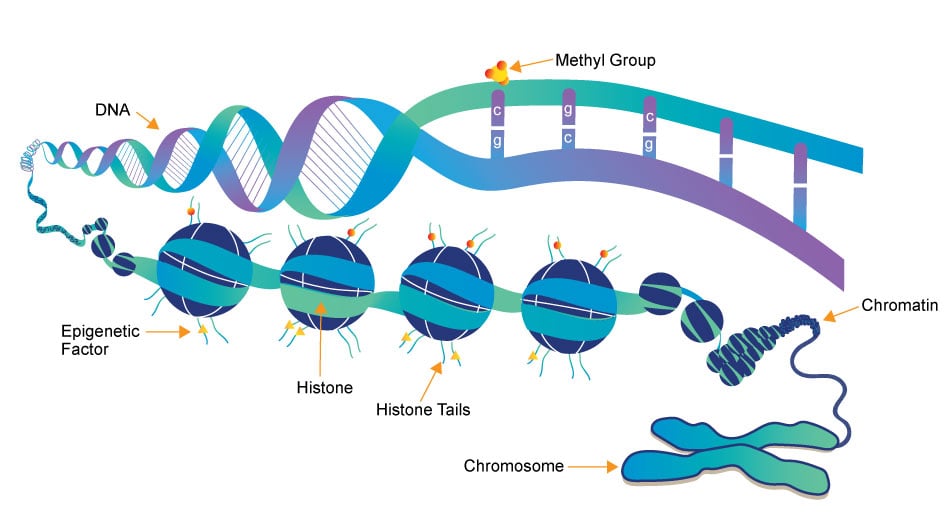
Epigenetics research delves into the molecular mechanisms that control gene expression and cellular traits without altering the underlying DNA sequence. One crucial aspect of this field is the role of small molecules, which act as powerful regulators of epigenetic modifications. These small compounds, typically comprising a few dozen to a few hundred atoms, have emerged as essential tools in understanding and manipulating the epigenome.
- DNA Methylation Inhibitors: Small molecules like 5-azacytidine and 5-aza-2'-deoxycytidine are DNA methyltransferase inhibitors. They block the addition of methyl groups to DNA, leading to DNA demethylation. This can reactivate silenced genes, potentially offering therapeutic avenues for conditions like cancer.
- HDAC inhibitors: HDACs remove acetyl groups from histone proteins, contributing to gene repression. Small molecule HDAC inhibitors, such as Vorinostat and Romidepsin, can reverse this process by increasing histone acetylation, allowing genes to be more accessible for transcription. These inhibitors are being explored for cancer therapy and other conditions.
- Histone Methyltransferase Inhibitors: Small molecules like GSK126 inhibit specific histone methyltransferases, affecting histone methylation patterns. This can alter gene expression, making them promising candidates for cancer and other diseases with epigenetic dysregulation.
- RNA Modulators: Small molecules can also target non-coding RNAs involved in epigenetic regulation. For instance, small molecules called small interfering RNAs (siRNAs) can be designed to target and degrade specific long non-coding RNAs, influencing gene expression.
- Epigenetic Reader Domain Inhibitors: These small molecules target proteins that recognize and bind to specific epigenetic marks. Examples include inhibitors of bromodomain-containing proteins (BET inhibitors), which can disrupt gene regulation by interfering with protein-DNA interactions.
Small molecules in epigenetics research not only provide insights into the fundamental biology of gene regulation but also hold immense promise for developing novel therapeutics. Their ability to selectively modulate specific epigenetic marks and pathways has led to ongoing clinical trials and drug development efforts for various diseases, including cancer, neurological disorders, and inflammatory conditions. Understanding and harnessing the power of these small molecules is at the forefront of modern epigenetics research, offering new hope for precision medicine and targeted therapies.
3 key components involved in the regulation of epigenetic modifications
Epigenetics Writer
Epigenetics writers are enzymes responsible for adding chemical marks or modifications to DNA or histone proteins. These marks include DNA methylation (addition of methyl groups to DNA) and histone modifications (such as acetylation, methylation, phosphorylation, etc.).
Epigenetics Reader
Function: Epigenetics readers are proteins that can recognize and bind to specific epigenetic marks on DNA or histones. These reader proteins interpret the epigenetic code and facilitate downstream cellular processes, such as gene activation or repression.
Epigenetics Eraser
Function: Epigenetics erasers are enzymes responsible for removing or reversing epigenetic marks on DNA or histones. This process allows for the dynamic regulation of gene expression and the resetting of epigenetic states during various stages of development and in response to environmental changes.
-
PARP1 Degrader
SK-575-NEG is a PARP1 degrader designed as a methylated counterpart of SK-575, synthesized through the methylation of the amino group in piperidine-2,6-dione. It demonstrates a strong binding affinity to PARP1, with an IC50 of 2.64 nM. However, SK-575-NEG does not induce PARP1 degradation in MDA-MB-436 and Capan-1 cell lines at concentrations up to 1 μM. This compound is valuable for research focusing on the mechanistic understanding of PARP1 inhibition and degradation pathways. -
PARP14 Inhibitor
PARP14 Inhibitor 2 is a highly selective inhibitor targeting PARP14 with an IC50 value of less than 30 nM. This compound effectively inhibits the mono-ADP-ribosyltransferase activity of PARP14, thereby modulating signaling pathways associated with IFN-γ and IL-4. By reversing protumor macrophage polarization and inhibiting pro-inflammatory responses, PARP14 Inhibitor 2 holds promise for the investigation of diseases related to PARP14, including tumors, atopic dermatitis, and autoimmune disorders. -
PARP1 Inhibitor
PARP1-IN-36 is a selective inhibitor of PARP-1, characterized as a 4-carboxamido-isoindolinone derivative with a Kd value of less than 0.01 μM. This compound exhibits significant biological activity in the modulation of cellular processes associated with DNA repair, making it a valuable tool in research focused on cancer, cardiovascular diseases, nervous system injury, and inflammation. Its potent inhibition of PARP-1 serves to elucidate the role of this enzyme in various pathological contexts. -
PARP-1 Inhibitor
6(5H)-Phenanthridinone is a potent inhibitor of PARP-1, a key enzyme involved in DNA repair processes. This compound exhibits significant immunomodulatory effects and has been shown to inhibit cell proliferation. It is employed in cancer research to explore therapeutic strategies targeting DNA repair pathways and to enhance the efficacy of chemotherapeutic agents. -
PARP1/2 Inhibitor
PARP1/2-IN-3 is a potent inhibitor of PARP1 and PARP2, exhibiting IC50 values of 0.2235 nM and <0.001 nM, respectively. This compound effectively inhibits the proliferation of Capan-1 wildtype and AZD2281 or BMN673-resistant cells, with IC50 values ranging from 1.82 to 9.98 nM. Additionally, PARP1/2-IN-3 demonstrates significant antitumor efficacy in murine models, making it a valuable tool for cancer research and therapeutic development targeting PARP pathways. -
CDK-1/PARP-1 Inhibitor
UNPD139734 is a potent inhibitor of Cyclin-Dependent Kinase 1 (CDK-1) and Poly (ADP-ribose) polymerase 1 (PARP-1), forming stable complexes with both target proteins. This compound serves as a valuable lead for the structural optimization of dual-target anticancer agents, particularly in the context of breast cancer research. Its dual inhibition mechanism offers a promising avenue for investigating novel therapeutic strategies in oncology. -
PARP1/2/7 Inhibitor
PARP-1/2/7-IN-1 is a highly potent inhibitor of poly (ADP-ribose) polymerases PARP-1, PARP-2, and PARP-7, demonstrating an IC50 of less than 10 nM. This compound is valuable for research applications focused on cancer biology, DNA repair mechanisms, and cellular stress responses. Its multifunctional inhibition can aid in the exploration of therapeutic strategies targeting PARP-mediated pathways in various diseases. -
PARP1 Inhibitor
PARP1-IN-48 is a potent and selective inhibitor of poly(ADP-ribose) polymerase 1 (PARP1), exhibiting an IC50 of 3 nM against PARP1 and 170 nM against PARP2. This compound is valuable for research in oncology, virology, and metabolic disorders, facilitating investigations into the role of PARP1 in DNA repair and cellular stress responses. Its selective action makes it an essential tool for studying therapeutic pathways in cancer treatment and other disease models. -
PARP1 Inhibitor
PARP1-IN-30 is a potent, selective inhibitor of PARP1 that exhibits cytotoxic effects in tumor cells. It effectively targets cancer cells with deficiencies in the breast cancer 1 protein (BRCA1) or BRCA2, offering a valuable tool for elucidating the role of PARP1 in DNA repair and cancer biology. This compound is particularly relevant for research applications focused on targeted cancer therapies and synthetic lethality in oncology. -
PARP Inhibitor
ARTD10/PARP10-IN-2 is a potent and non-selective inhibitor of the poly(ADP-ribose) polymerases, specifically targeting ARTD10/PARP10 and ARTD1/PARP1. With IC50 values of 2.0 μM and 9.7 μM, respectively, this compound effectively modulates mono-ADP-ribosyltransferase and poly(ADP-ribose) polymerase activities. Its ability to inhibit these pathways makes ARTD10/PARP10-IN-2 valuable for research on DNA repair mechanisms and therapeutic approaches in cancer treatment. -
PARP1 Inhibitor
ZINC000081009201 is a potent inhibitor of poly(ADP-ribose) polymerase 1 (PARP1) with an IC50 value of 1.4767 μM. This compound demonstrates significant potential for the study of triple-negative breast cancer (TNBC) by targeting PARP1-mediated repair pathways. Its inhibition may aid in elucidating the mechanisms of resistance and sensitivity in cancer treatment, making it a valuable tool for cancer research applications. -
PARP7 Inhibitor
PARP7-IN-18 is a potent selective inhibitor of PARP7, exhibiting an IC50 value of 0.11 nM. This compound demonstrates significant anticancer activity, making it a valuable tool for research applications focused on cancer biology and the therapeutic potential of targeting PARP7. Its favorable pharmacokinetic properties further enhance its utility in in vivo studies and drug development. -
PARP Inhibitor
NU 1085 is a potent poly(ADP-ribose) polymerase (PARP) inhibitor with an impressive Ki of 6 nM. This compound exhibits significant cytotoxicity towards cancer cells, with an LC50 between 83-94 μM. Additionally, NU 1085 has the potential to enhance the anticancer effects of Temozolomide, making it a valuable tool for cancer research, particularly in the study of lung cancer and other malignancies. -
PARP1 Inhibitor
PARP1-IN-20, a selective PARP1 inhibitor, exhibits a potent inhibitory effect with an IC50 of 4.62 nM. This compound demonstrates minimal PARP-Trapping activity compared to other known inhibitors, including a significant threshold of >100 μM in the MDA-MB-436 cell line. PARP1-IN-20 is suitable for research applications focused on cancer therapy and DNA damage repair mechanisms. -
Conjugate Compound with PARP Inhibitor
ADP-ribose/PARP-IN-1 is a conjugated compound that combines disease-targeting moieties with PARP inhibitor functionality. This reagent selectively delivers PARP inhibitors to tumor cells, facilitating the inhibition of PARP enzymes that are critical for DNA damage repair. The presence of a cleavable linker ensures the release of the PARP inhibitor under specific conditions, while the chelator component enables the accumulation of radionuclides that exert cytotoxic effects. ADP-ribose/PARP-IN-1 is a valuable tool for research into prostate cancer and other malignancies characterized by PARP dependency. -
PARP Inhibitor
LT-626 is a potent poly(ADP-ribose) polymerase (PARP) inhibitor, with an IC50 of 1.60 nM. This inhibitor effectively reduces cellular poly(ADP-ribose) synthesis, demonstrating an EC50 of 17.9 nM, and displays enhanced cytotoxicity in colorectal cancer cells with MRE11 mutations. Additionally, LT-626 exhibits synergistic effects when combined with chemotherapeutic agents such as Cisplatin, Oxaliplatin, and SN-38, making it a valuable tool for colorectal cancer research applications. -
PARP7 Inhibitor
RBN010860 is a potent inhibitor of PARP7, exhibiting an IC50 value of less than 0.1 μM. This reagent serves as a valuable tool for investigating the role of PARP7 in cancer biology, enabling researchers to explore its potential as a therapeutic target in oncological studies. -
PARP7 Inhibitor
PARP7-IN-12 is a potent inhibitor of PARP7, exhibiting an IC50 value of 7.836 nM. This compound demonstrates significant potential in cancer research by modulating poly(ADP-ribose) polymerase activity, which is associated with DNA damage response pathways. PARP7-IN-12 may serve as an effective tool for elucidating the role of PARP7 in tumor biology and for developing therapeutic strategies targeting PARP-related pathways in cancer. -
PARP Inhibitor
PARP1-IN-6 is a dual inhibitor of tubulin and PARP-1, displaying IC50 values of 0.94 μM and 0.48 μM, respectively. This compound demonstrates significant biological activity by disrupting cellular processes mediated by both targets, making it a valuable tool for cancer research. PARP1-IN-6 is particularly useful in studies investigating the roles of PARP inhibition in DNA repair mechanisms and cytoskeletal dynamics. -
PARP1 Inhibitor
DPQ hydrochloride is a potent and selective inhibitor of PARP-1 (poly(ADP-ribose) polymerase 1), effectively blocking PARP-1-mediated DNA damage repair and reducing NAD+/ATP consumption. This compound demonstrates significant anti-inflammatory properties by inhibiting the activation of the NF-κB pathway, leading to a decrease in pro-inflammatory cytokines such as TNF-α and IL-6, as well as mitigating oxidative stress. DPQ hydrochloride is ideal for research applications related to inflammation and can be utilized in studies of conditions such as acute lung injury, myocardial infarction, and neurodegenerative diseases. -
PARP-1 Inhibitor
Lotixparib is a potent inhibitor of poly(ADP-ribose) polymerase-1 (PARP-1), a key enzyme involved in DNA damage repair. This compound demonstrates cytoprotective effects, offering potential therapeutic applications in retinal diseases. Lotixparib is valuable for research focused on PARP-1-related conditions and mechanisms underlying DNA repair pathways. -
PARP1 PROTAC-type Degrader
Vrucaparib-TP4 is a PROTAC-type degrader that targets PARP1. It facilitates the ubiquitination and subsequent degradation of PARP1, making it a valuable tool in cancer research. This compound is particularly useful for investigating the role of PARP1 in tumor biology and evaluating therapeutic strategies that exploit the degradation pathway for potential anti-tumor effects. -
PARP7 Inhibitor
PARP7-IN-23 is a potent inhibitor of Poly(ADP-ribose) polymerase 7 (PARP7) with an EC50 of 0.915 nM for phosphorylated STAT1 (pSTAT1) in NCI-H1373 cells. This compound demonstrates significant inhibitory activity, which positions it as a valuable tool for cancer research, particularly in studies involving STAT1 signaling pathways and therapeutic interventions targeting PARP7. -
PARP1 Inhibitor
PARP1-IN-21 is a potent inhibitor of PARP1, exhibiting an IC50 of less than 10 nM. This compound effectively disrupts the poly(ADP-ribose) polymerase 1 enzymatic activity, leading to the inhibition of DNA repair processes. It is primarily utilized in research applications focused on cancer biology and therapeutic development, particularly in the exploration of combination therapies for tumors with defective homologous recombination. -
EZH2 PROTAC Degrader
PROTAC EZH2 Degrader-44 is a selective degrader targeting the EZH2-PRC2 complex through the recruitment of the CRBN E3 ligase, facilitating proteasome-mediated degradation of EZH2, SUZ12, and EED. This process leads to a marked reduction in H3K27me3 and CARM1 levels, resulting in potent antiproliferative effects. It induces mitochondrial dysfunction and promotes apoptosis by modulating Bcl-2 family proteins, demonstrating minimal cytotoxicity in normal human mammary epithelial, liver, and kidney cells. PROTAC EZH2 Degrader-44 serves as an effective research tool for investigating targeted therapies in triple-negative breast cancer. -
PARP1 Inhibitor
PARP1-IN-43 is a potent PARP1 inhibitor with an IC50 of 5 nM, exhibiting effective penetration across the blood-brain barrier. This compound is primarily utilized in research focused on homologous recombination-deficient central nervous system (CNS) tumors, facilitating the exploration of targeted therapies in oncology. Its ability to selectively inhibit PARP1 makes it a valuable reagent for investigating DNA repair mechanisms and their implications in cancer biology. -
PARP1 Inhibitor
PARP1-IN-39 is a potent inhibitor of PARP1, exhibiting an IC50 of 0.22 nM, and an IC50 of 1.57 nM in human breast cancer cells. This compound is valuable for investigating cancers associated with DNA repair deficiencies, including those linked to BRCA1/2 mutations, and is applicable in studies of breast, ovarian, pancreatic, and prostate cancers. PARP1-IN-39 facilitates research into targeted therapies that exploit the vulnerabilities of cancer cells with defective DNA repair mechanisms. -
PARP Inhibitor
KCL-440 is a potent PARP inhibitor that effectively penetrates the central nervous system, exhibiting an IC50 of 68 nM for PARP-1 inhibition. This compound is primarily utilized in research exploring DNA repair mechanisms, cancer therapy, and neurodegenerative disease models, making it a valuable tool for studying therapeutic strategies targeting cellular stress responses. -
PARP7 Probe
PARP7-probe-1 is a biotinylated chemiluminescent probe specifically designed to bind to the active site of PARP7. This reagent allows for the investigation of PARP7's enzymatic activity and its role in various biological processes. PARP7-probe-1 is suitable for applications involving protein interactions and the study of cellular mechanisms associated with PARP7 function. -
PARP7 Inhibitor
TIPARP-IN-1 is a selective inhibitor of PARP7 (TIPARP) with an IC50 of 2.15 nM. By inhibiting TIPARP, this compound restores the interferon signaling pathway in tumors, effectively enhancing the anti-tumor immune response within the tumor microenvironment while minimizing systemic cytokine production. TIPARP-IN-1 is an important tool for investigating the role of PARP7 in head and neck squamous cell carcinoma. -
PARP Inhibitor
4-Aminonaphthalimide is a potent inhibitor of poly(ADP-ribose) polymerase (PARP), a key enzyme involved in DNA repair processes. This compound enhances the cytotoxic effects of γ-radiation in cancer cells, making it a valuable tool for studies focused on cancer therapy and radiation biology. Its dual function as a PARP inhibitor and radiation sensitizer positions 4-Aminonaphthalimide as an important reagent for research applications in oncology and therapeutic resistance. -
PARP1 Inhibitor
PARP1-IN-51 is a potent inhibitor of the enzyme PARP1, which plays a critical role in DNA repair mechanisms. This compound exhibits significant biological activity by disrupting the PARP1-mediated repair of single-strand breaks, ultimately leading to cancer cell apoptosis. PARP1-IN-51 is particularly valuable for research in oncology, including studies focused on breast cancer and other malignancies with defective DNA repair pathways. -
PARP1/2 Inhibitor
PARP1-IN-37 is a selective inhibitor of poly(ADP-ribose) polymerases 1 and 2 (PARP1/2), exhibiting an IC50 of 24 nM for PARP1. This compound effectively inhibits PARP activity in cellular assays with an EC50 of 3.7 μM. PARP1-IN-37 is particularly relevant for investigations into BRCA-mutated tumors, including breast and ovarian cancers, making it a valuable tool for cancer research and therapeutic development. -
PARP1 Inhibitor
PARP1-IN-18 is a potent inhibitor of the poly(ADP-ribose) polymerase 1 (PARP1) enzyme, demonstrating an IC50 value of 2.7 nM. This compound exhibits significant anticancer activity, making it a valuable tool for research applications targeting DNA repair mechanisms and cancer therapeutics. PARP1-IN-18 can be utilized to explore PARP-mediated cellular processes and evaluate its potential in combination therapies for various malignancies. -
PARP7 Inhibitor
PARP7-IN-24 is a highly potent inhibitor of PARP7, exhibiting an EC50 of 0.375 nM for pSTAT1 in NCI-H1373 cells. This compound is poised for significant contributions to cancer research, facilitating the study of PARP7's role in tumor biology and potential therapeutic interventions. Its strong inhibitory activity makes it a valuable tool for exploring the molecular mechanisms underlying cancer progression and treatment responses. -
PARP-1 Inhibitor
PARP-1-IN-23 is a selective inhibitor of poly(ADP-ribose) polymerase 1 (PARP-1), exhibiting an IC50 of 12.38 nM. This compound demonstrates significant antitumor activity in vivo, making it a valuable tool for studying the mechanisms of cancer progression and treatment. Its inhibition of PARP-1 suggests potential applications in enhancing the efficacy of chemotherapy and targeting cancer cell survival pathways. -
PARP1/2 Inhibitor
Senaparib hydrochloride is a selective inhibitor of PARP1 and PARP2, demonstrating significant anti-tumor activity. This compound is particularly effective in the treatment of advanced ovarian cancer, making it relevant for research focused on cancer therapeutics and mechanisms of tumor progression. Its ability to interfere with DNA repair pathways positions Senaparib hydrochloride as a valuable tool in understanding the efficacy of PARP inhibition in oncology. -
PARP Inhibitor
PARP-2/1-IN-2 is a potent inhibitor of poly (ADP-ribose) polymerases 1 and 2, exhibiting Kis of 2 nM and 5 nM, respectively. It demonstrates a biological activity with an EC50 of 3 nM in cell-based assays assessing PARP activity. This compound is valuable for research focused on cancer therapeutics, DNA repair mechanisms, and the role of PARP in various cellular processes. -
PARP Inhibitor
PARP1-IN-28 is a potent inhibitor of Poly(ADP-ribose) polymerase 1 (PARP1), a crucial enzyme involved in DNA repair and cellular response to stress. This compound exhibits significant anti-proliferative activity and is particularly relevant in cancer research, enabling studies on tumor cell viability and resistance mechanisms. Its application extends to assessing the efficacy of combination therapies in oncology, particularly in contexts involving homologous recombination deficiency. -
PARP1/2 Inhibitor
PARP1/2-IN-4 is a selective inhibitor of PARP1 and PARP2 enzymes, which play crucial roles in DNA repair processes via the base excision repair pathway. This compound exhibits significant potential in preclinical research for cancer therapy by enhancing the efficacy of DNA-damaging agents and promoting synthetic lethality in tumors with deficient DNA repair mechanisms. It is a valuable tool for studying DNA repair pathways and evaluating therapeutic strategies in oncology research. -
PARP10 Inhibitor
PARP10-IN-2 is a selective inhibitor of the mono-ADP-ribosyltransferase PARP10, exhibiting an IC50 of 3.64 µM in human PARP10 assays. This compound also effectively inhibits PARP2 and PARP15, with IC50 values of 27 µM and 11 µM, respectively. PARP10-IN-2 is valuable for research applications focusing on cellular processes influenced by ADP-ribosylation and has potential implications in cancer and neurodegenerative disease studies. -
PARP7 Inhibitor
KMR-206 is a selective inhibitor of PARP7, demonstrating an IC50 of 13.7 nM. This compound enhances the activation of STAT1 and phosphorylated STAT1, thereby promoting type I interferon signaling through STING degradation. KMR-206 exhibits notable anticancer activity, particularly against lung adenocarcinoma, and serves as a valuable tool for investigating its effects in colon cancer research. -
PARP Inhibitor
YCH1899 is an orally active PARP inhibitor, demonstrating a potent IC50 of less than 0.001 nM for PARP1/2. This compound exhibits significant antiproliferative effects against Capan-1 cells resistant to Olaparib and Talazoparib, with IC50 values of 0.89 nM and 1.13 nM, respectively. YCH1899 also presents favorable pharmacokinetic properties in rat models, making it a valuable tool for research in cancer therapeutics and resistance mechanisms. -
PARP-1 Inhibitor
PARP1-IN-29 is a potent PARP-1 inhibitor with an IC50 of 6.3 nM, demonstrating strong efficacy in modulating DNA damage repair pathways. Labeled with [18F], this compound is suitable for positron emission tomography (PET) imaging, allowing for targeted visualization of PARP-1 activity in tumors. Its applications extend to oncology and imaging research, providing valuable insights into cancer biology and treatment response. -
PARP-1 Inhibitor
5-AIQ (5-Aminoisoquinolin-1-one) is a selective inhibitor of PARP-1, a key protein involved in DNA repair mechanisms. This compound exhibits significant potential in reducing tissue injury resulting from ischemia-reperfusion of the liver. 5-AIQ is useful for research applications focused on understanding ischemia-reperfusion injury and developing therapeutic strategies for liver protection. -
PARP Inhibitor
UKTT15 is an allosteric inhibitor of the enzyme PARP1. It exhibits significant inhibitory activity against PARP1, influencing DNA repair mechanisms and cellular response to DNA damage. UKTT15 is useful in research focused on cancer biology, particularly in studies evaluating the therapeutic potential of PARP inhibition in cancer treatment and the modulation of DNA repair pathways. -
PARP-1/ARTD-1 Inhibitor
EB-47 is a selective inhibitor of PARP-1 (ARTD-1) with an IC50 value of 45 nM, demonstrating its strong inhibition profile. It also exhibits modest potency against ARTD5 with an IC50 of 410 nM. Structurally, EB-47 mimics the substrate NAD+, extending from the nicotinamide to the adenosine subsite, making it a valuable tool for studying DNA repair mechanisms and related cancer research applications. -
PARP Inhibitor
AZ3391 is a selective inhibitor of the PARP enzyme family, which is critically involved in cellular processes including DNA repair, chromatin remodeling, and cell cycle regulation. As a quinoxaline derivative, AZ3391 demonstrates significant potential for investigating neurodegenerative diseases and conditions affecting the central nervous system, particularly in the brain and spinal cord. This compound is valuable for research applications focused on understanding the mechanisms underlying DNA damage response and related pathologies. -
PARP-1 Inhibitor
Amelparib is a selective inhibitor of PARP-1, exhibiting potent activity with an IC50 of 18.5 nM for PARP-1 inhibition and 10.7 nM for cellular PAR formation. This orally active and water-soluble compound demonstrates potential neuroprotective effects, making it a valuable tool for research in the field of acute ischemic stroke. Amelparib's ability to modulate PARP-1 activity offers insights into its therapeutic roles in neuroprotection and cellular repair mechanisms. -
PARP Inhibitor
PARP1-IN-7 is a selective inhibitor of poly(ADP-ribose) polymerase-1 (PARP1), primarily functioning to disrupt DNA repair mechanisms in cancer cells. By inhibiting PARP1 activity, it enhances the cytotoxic effects of DNA-damaging agents, making it a valuable tool in cancer research. Its applications include studying mechanisms of tumorigenesis, evaluating synthetic lethality in cancer therapies, and exploring potential combinations with other anticancer agents.
